Abstract
Comment on: Roberti et al. Cell Cycle 2011; 10:127–34.
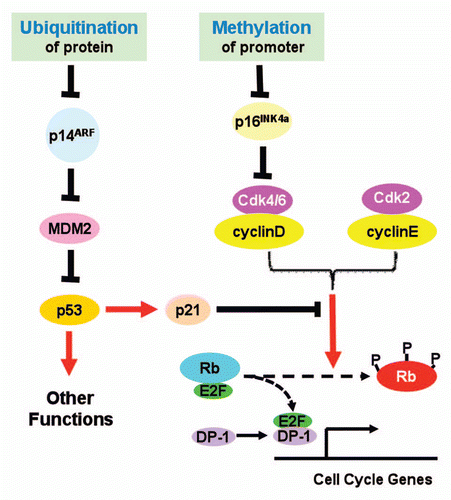
The INK4b/INK4a/ARF locus encodes three tumor suppressors: p15INK4b, p16INK4a and p14ARF.Citation1 These three proteins play important roles in cell cycle control and tumor suppression. Both p15INK4b and p16INK4a serve as inhibitors of cyclinD-Cdk4/6 activity, and prevent the Rb family tumor suppressors from hyperphosphorylation, thus repress E2F-transactivated cell cycle genes. p14ARF antagonizes MDM-mediated ubiquitination and subsequent degradation of p53. As a transcription regulator, p53 transcriptionally promotes cell apoptosis and growth arrest, thus functions as a tumor suppressor. ARF also has been reported to suppress tumor growth by p53-independent pathway.Citation2
Given the importance of the INK4b/INK4a/ARF locus, mechanisms underlying its regulation in normal cells, and more importantly, its inactivation in cancer cells have been intensively studied. One established mechanism that silences the whole locus involves CDC6, which may represent the coordinated control of DNA replication and transcriptional repression during cell division.Citation3 Genetic alterations including deletion and missense mutations have been reported in a variety of tumors. Interestingly, INK4a and ARF, each has a unique promoter and exon 1, share the other two exons but using alternative reading frames. This genetic architecture increases the complexity of individualized regulation of expression. However, it has been known that some stimuli may specifically regulate either p16INK4a or p14ARF. Promoter-specific methylation has been reported to silence either INK4a or ARF.Citation4
In a previous issue of Cell Cycle, Roberti et al. provided data to suggest another layer of regulation of INK4a/ARF locus in Burkitt's lymphoma cell lines ().Citation5 They reported that in those cell lines the promoter of INK4a was heavily methylated but that of ARF was not. Accordingly, they found that the mRNA levels of INK4a were ubiquitously down-regulated whereas those of ARF, up-regulated. These up-regulated levels of ARF mRNA, however, apparently failed to result in elevated levels of p14ARF proteins. To explain this discrepancy, the authors explored the protein turnover in those cell lines. They were able to show that inhibition of proteasomal activity by incubating cells with MG132, a well known inhibitor of proteasomes, increased the protein levels of p14ARF. Furthermore, ubiquitinated forms of p14ARF were detected in protein samples from MG132-treated cells. Taken together, these data provided strong evidence to support that in Burkitt's lymphoma cell lines used in this study, INK4a was mainly repressed by promoter methylation, whereas p14ARF may be down-regulated by accelerated degradation by the ubiquitination-proteasome system.
As perhaps all other excellent studies, this interesting one also raises more questions than it has answered. Since p14ARF lacks lysyl residue, its ubiquitination has been reported to be mediated by the N-terminal α-amino group, instead of the more commonly reported ε-amino group of lysyl residues.Citation6 For p14ARF is mainly localized in nucleolus and is stabilized by its interaction with NPM/B23,Citation7 its degradation by the proteosomes is slow in most cell lines. It would be interesting to explore the molecular and biochemical mechanisms underlying this cell type-specific instability of p14ARF in those Burkitt's lymphoma cell lines. A particularly interesting question would be if this accelerated decay results from a mutation-driven p14ARF misfolding or disruption of its interaction with NPM/B23. It is also possible that a mutation of NPM/B23 may alter p14ARF function and subcellular localization. In addition, ubiquitination-independent degradation of regulatory proteins such as HIF-1α, p53 and p27 in tumor cells can be triggered by various chemotherapeutics or other stresses.Citation8,Citation9 While the ubiquitination of p14ARF was demonstrated, an interesting question would be whether such ubiquitination is a bona fide prerequisite for p14ARF degradation, or simply a consequence of accumulation of p14ARF when proteasomal activity was blocked. Future investigations stimulated by this report surely will significantly advance our understanding of the regulation of p14ARF and growth suppression.
In conclusion, these interesting new findings, together with published data from other researchers, depict an updated view of the regulation of tumor suppressive function of this locus. Both promoter methylation and accelerated ubiquitination may play roles in individualized control of INK4a and ARF expression, at least, in those Burkitt's lymphoma cell lines. The insight and perspectives brought by this new study may facilitate the identification of novel drug targets for the development of novel cancer therapy.
Figures and Tables
Figure 1 Proposed roles of protein ubiquitination and promoter methylation in control of INK4a/ARF expression. See text for detailed explanation.
Breast cancer is one of the most common cancers among women worldwide. Cyclin D1 and cyclin E are frequently overexpressed in breast cancers.Citation1 Cyclin D1 is overexpressed in ∼45% of breast cancers, and cyclin E is overexpressed in ∼30% of breast cancers.Citation1 Cyclin D or cyclin E overexpression is often found in aggressive breast cancers.Citation1 Several studies have shown that overexpression of cyclin E or amplification of the cyclin D1 gene is associated with a poor outcome.Citation1
Smad3, which is essential for mediating TGF-β antiproliferative response, is phosphorylated by cyclin D-CDK4 and cyclin E-CDK2.Citation2–Citation4 CDK phosphorylation of Smad3 inhibits its transcriptional activity and contributes to G1 cell cycle arrest.Citation2–Citation3 Several lines of evidence suggest that Smad3 plays an important role in the inhibition of breast tumorigenesis. For instance, TGF-β-induced growth inhibition and apoptosis is essentially lost in Smad3-/- mammary epithelial cells.Citation5 Overexpression of Smad3 in a breast cancer cell line causes cell cycle arrest accompanied with increased p15 levels, decreased cyclin A expression, and inhibition of Rb phosphorylation; conversely, overexpression of a dominant negative Smad3 accelerated cell proliferation. Furthermore, introduction of Smad3 into a breast cancer cell line reduced its tumorigenicity.Citation6 Low levels of nuclear Smad3 are associated with high-grade tumor, large tumor size, and estrogen receptor-negative tumors in human patients. c-myc is overexpressed in ∼38% breast cancers.Citation1 Smad3 plays a key role in mediating downregulation of c-myc in response to TGF-β, and this response is lost in breast cancer cell lines.Citation7
To address the question whether inhibition of Smad3 activity contributes to the oncogenic effects of cyclin E overexpression in breast cancer, Cooley et al. introduced the wild-type Smad3 or Smad3 with mutations at the CDK phosphorylation sites into the parental, vector control, or cyclin E-overexpressing (EL1) MCF7 breast cancer cell line.Citation8 MCF7 cells were used as they contain increased cyclin E levels. Smad3 containing CDK phosphorylation site mutations led to higher p15 transcript levels and lower c-myc transcript levels than the wild-type Smad3, with the greatest effect in the cyclin E-overexpressing cells. In transient Smad3-responsive reporter gene assays, overexpression of cyclin E repressed the wild-type Smad3 transcriptional activity, resulting in the CDK phosphorylation mutant Smad3 having a much higher activity than the wild-type Smad3. Moreover, cotransfection with a CDK2 siRNA or treatment with a CDK2 inhibitor led to a higher Smad3 reporter gene activity than the control cells. Thus, cyclin E-overexpression is associated with phosphorylation and inhibition of Smad3 activity by cyclin E-CDK2 in breast cancer cells. In a similar study, Zelivianski et al. showed that cyclin D-CDK4 inhibits Smad3 activity in cyclin D1-overexpressing breast cancer cells.Citation9
Therapeutics targeting CDKs or cyclins hold promise for subsets of breast cancers. For example, Her2 activates cyclin D1 expression. Ablation of cyclin D1 protects against Her2-induced breast tumorigenesis in animal models.Citation1 In human breast cancer, approximately 25% of Her2-overexpressing tumors contain high levels of cyclin D1. This subset of patients may benefit from inhibiting CDK4 kinase.Citation1 For the ER-positive postmenopausal breast cancer patients, a subset of them overexpress the low molecular weight forms of cyclin E, which are derived mainly from the full length cyclin E by posttranslational processing and result in higher CDK2 activity.Citation10 This subset of patients is associated with a poor prognosis and more likely to develop resistance to endocrine therapy that causes G1 cell cycle arrest.Citation10 The study further showed that treatment of breast cancer cells with a CDK2 inhibitor can reverse the resistance.Citation10 These observations support clinical investigations of CDK2 inhibitor therapy for this subset of breast cancer patients. These potential therapies are expected to restore Smad3 tumor suppressive function. Furthermore, inhibition of CDK-mediated phosphorylation of Smad3 not only restores its tumor suppressive function, but also is expected to eliminate its invasive function.Citation4 Future studies in these important areas are fully anticipated.
References
- Butt AJ, et al. Adv Exp Med Biol 2008; 630:189 - 205
- Matsuura I, et al. Nature 2004; 430:226 - 231
- Li L, et al. Nat Immunol 2006; 7:1157 - 1165
- Matsuzaki K, et al. Cancer Res 2009; 69:5321 - 5330
- Kohn EA, et al. Breast Cancer Res 2010; 12:R83
- Tian F, et al. Cancer Res 2004; 64:4523 - 4530
- Chen CR, et al. Proc Natl Acad Sci USA 2001; 98:992 - 999
- Cooley A, et al. Cell Cycle 2010; 9:4900 - 4907
- Zelivianski S, et al. Mol Cancer Res 2010; 8:1375 - 1387
- Akli S, et al. Clin Cancer Res 2010; 16:1179 - 1190
Over the past several decades, the TGF-β pathway has simultaneously captivated and confused scientists. TFG-α binding to cell-surface receptors induces a complex circuit of signaling events with diverse targets of gene regulation; dissection of the effects of these downstream events are further complicated by context dependent mechanisms. Indeed, one of the most intriguing aspect of the TGF-β axis is the ability to serve as both a tumor suppressor and a pro-tumorigenic factor depending on the stage of cancer development1. In a previous issue of Cell Cycle, work from Cooley and colleagues suggest that CDK2 plays a critical role in tempering the anti-proliferative activity of the TGF-β pathway.Citation9
Upon ligand binding, TGFαRII phosphorylates and activates TGRFαRI, a serine/threonine kinase responsible, in turn, for phosphorylation of Smad2 and Smad3 (R-Smads). Ultimately, phosphorylated Smad2/3 translocates to the nucleus and associates with Smad4 where they trigger increased expression of a number of key genes including anti-proliferative factors such as p15ink4b and p21Cip1.Citation2,Citation3 The resulting effect is net inhibition of the cell cycle components Cdk4 and Cdk2, respectively. Furthermore, activated Smads also transcriptionally repress the potent oncogene c-Myc, among other growth-related targets.Citation4 Thus, Smad-dependent signaling through TGF-β provides robust anti-proliferative influences.
While TGF-β/Smad activates p15ink4b and p21Cip1 to inhibit cell cycle progression, Cdk2 (and Cdk4) can also phosphorylate Smad3 at multiple sites distinct from those targeted by TGFαRI.Citation5 In contrast to receptor-mediated phosphorylation, Cdk2-dependent phosphorylation is inhibitory to Smad3 function;Citation6 perturbation of Cdk2 activity or mutation of these phosphorylation sites leads to increased Smad3-dependent p15ink4b/Myc transcriptional regulation. These findings directly link cyclin E/Cdk2 with the inhibition of the Smad anti-proliferative pathway of the TGF-β axis and suggest that TGF-β/Smad3 and cyclin/Cdk signals provide opposing influences on proliferation. The current work of Cooley et al. demonstrate that indeed cyclin E overexpression in breast cancer leads to attenuation of Smad3 transcriptional activity towards anti-proliferative mediators. In human breast cancer cells, the target genes p15ink4b, p21cip1, and c-Myc were regulated in the presence of ectopically expressed Smad3 and this regulation of was attenuated by increased cyclin E expression. Critically, targeted mutations in documented Cdk2 phosphorylation sites of Smad3 abrogated regulation, as did pharmacologic inhibition of Cdk2, providing a direct link between Cdk2 and Smad3.
Paradoxically, significant literature also reveals a role for TGF-β/Smad3 in promoting tumorigenesis, which has been demonstrated through several mechanisms.Citation7 First, Smad3 mediates TGF-β dependent immune suppression. Immune-related targets of Smad3 signaling include MHC class II, interferon-gamma, and interleukins. Loss of Smad3 may impair tumor cells ability to escape immune surveillance. Second, Smad3 signaling contributes directly to TGF-β induced epithelial-mesenchymal transition by acting as a transcriptional activator of Snail, a repressor of E-cadherin. Cells with a dysfunctional TGF-β pathway, such as those derived from Smad3-deficient mice, fail to induce EMT. Finally, Smad3 activity has been positively correlated with metastatic potential and angiogenic responses through regulation of factors such as MMPs and CTGF. Thus, because Smad3 protects against and later also facilitates tumor progression, it may be advantageous for malignant cells to suppress Smad3 activity reversibly; upon development of resistance to the downstream anti-proliferative effects of TGF-β/Smad3, cancer cells may harness Smad3 to promote tumor growth and metastatic potential.
Consistent with this idea, Smad3 is rarely mutated in human cancer. Instead, in several cancers, such as breast and gastric, Smad3 is often subjected to down-regulation at the protein level. Furthermore, attempts to model Smad3 deficiency in the mouse have met with conflicting results,Citation7 perhaps a testament to the dual role for Smad3 in tumor suppression and oncogenesis. Taken together, these observations suggest that dynamic regulation of Smad3 protein expression/activation could be an important determinant of cancer fate.
Cooley et al. investigate cyclin E/Cdk2 regulation of Smad3-dependent transcription, seeking to uncover the mechanism by which breast cancers over-expressing Cyclin E correlate with aggressive phenotypes and carry poor prognosis.Citation8 The authors provide evidence that overexpressed cyclin E, through the Cdk2 kinase activity, neutralizes the anti-proliferative signaling of Smad3. Future studies might address whether the inhibitory Cdk phosphorylation of Smad3 represses its function universally or only conditionally. That is, given the diverse biological functions mediated by Smad3 targets, it is reasonable to suspect that phosphorylation of Smad3 by Cdks specifies activity towards a subset of downstream genes. If the pro-tumorigenic effects of Smad3 transcriptional regulation, eg. targets such as Snail and MMPs, are un-perturbed Cdk phosphorylation, it would suggest a very intriguing mechanistic clue as to why Smad3 is epigenetically regulated and seldom mutated in human cancer.
References
- Massague J. Cell 2008; 134:215 - 230
- Moustakas A, et al. Immunol Lett 2002; 82:85 - 91
- Ten Dijke P, et al. J Cell Physiol 2002; 191:1 - 16
- Yagi K, et al. J Biol Chem 2002; 277:854 - 861
- Matsuura I, et al. Nature 2004; 430:226 - 231
- Liu F, et al. Cell Cycle 2005; 4:63 - 66
- Millet C, et al. Crit Rev Eukaryot Gene Expr 2007; 17:281 - 293
- Keyomarsi K, et al. N Engl J Med 2002; 347:1566 - 1575
- Cooley A, et al. Cell Cycle 2010; 9:4900 - 4907
In most tissues, metabolism operates according to the “bend-but-don't-break” principle. Homeostatic mechanisms allow cells to respond to changes in nutrient availability or workload, thereby matching metabolic supply and demand. The end result is that cells can survive and maintain function during fluctuations in the availability of preferred nutrients like glucose. A classic example is the enhanced β-oxidation of fatty acids that occurs in muscle during energy deprivation or increased energy demand.Citation1 This transition is regulated by the fuel sensor AMP-activated protein kinase (AMPK), which shifts the metabolic balance from anabolic, energy-consuming activities towards catabolic, energy-generating ones.
In cancer cells, transforming mutations can limit metabolic flexibility and lead to an inescapable requirement (“addiction”) for specific nutrients. These abnormally rigid metabolic networks prevent cells from surviving acute interruptions in nutrient availability. Constitutive activation of PI3K/Akt signaling addicts tumor cells to glucose by interfering with the induction of fatty acid oxidation when glucose is withdrawn.Citation2 c-Myc addicts cells to glutamine by preventing them from supplying the tricarboxylic acid cycle using other nutrients.Citation3 Thus nutrient addiction is often cited as a basis for metabolic therapy in cancer. The essential premise: even though most tissues prefer to use glucose, glutamine, or other abundant nutrients to supply essential metabolic functions, systemic inhibition of these activities might preferentially kill malignant cells that cannot normally adapt by engaging compensatory pathways.
But other work suggests that metabolic rigidity is not a universal feature of malignant transformation. A subset of enzymes, particularly glutamate dehydrogenase, can enable intensely glycolytic tumor cells to survive inhibition of glucose metabolism.Citation4,Citation5 Some mutations in KRAS or BRAF provide colorectal cancer cells with a growth advantage in low-glucose conditions.Citation6 And now, in a previous issue of Cell Cycle, Chen and Shtivelman provide evidence for another mechanism of metabolic plasticity in cancer cells. They show that the protein CC3/TIP30 commits cells to glycolytic metabolism, and when its expression is chronically suppressed (as observed in tumors), cells achieve the ability to resist glucose deprivation.Citation7
Cells derive energy from glycolysis, oxidative phosphorylation or both. In glycolysis, ATP is produced very rapidly in the cytosol, although the yield of ATP per glucose is low. In oxidative phosphorylation, oxidation of various nutrients (fatty acids, amino acids, glucose and other sugars) in the mitochondria results in the delivery of reducing equivalents to the electron transport chain, producing a maximal yield of ATP. Chen and Shtivelman find that CC3, a tumor suppressor,Citation8 limits cellular ability to survive culture in low glucose. When the authors experimentally suppressed CC3 expression in HeLa and MCF7 cells to mimic levels observed in tumors, the modified cells displayed improved viability when challenged with low glucose. They were also better at sustaining their ATP levels than CC3-expressing cells. This difference may have been the result of an augmented capacity for oxidative metabolism, because cells with silenced CC3 had higher oxygen consumption and higher expression of electron transport chain subinits during prolonged low-glucose culture. Bioenergetic stability in low glucose was also suggested by diminished activation of AMPK. Thus CC3 suppression appears to result in a state of enhanced metabolic agility, allowing cancer cells to protect their energy stores in the face of limited access to glucose. More work is needed to understand how CC3, which lacks an obvious role in metabolism, influences nutrient dependence. It is interesting that CC3 is also proposed to suppress metastasisCitation9—surely metabolic agility would be advantageous as cells attempt to survive the rigors of matrix detachment, migration to a remote site, and establishment of a new focus of tumor growth.
Where tumor cells reside along the spectrum of metabolic flexibility/rigidity may well determine their sensitivity to therapy. This issue is more than an intellectual exercise related to the dream of future therapies directed against nutrient utilization. Many agents already in use likely impact tumor metabolism by interfering with the signaling pathways that regulate metabolic flux and flexibility. Cells with reduced levels of CC3 might be quite successful at resisting the metabolic stress brought on by targeting PI3K, mTOR, Bcr-Abl and other nodes of signal transduction. Forcing these cells to implement a more rigid metabolic platform could be a key factor in causing them to “break” rather than “bend.”
References
- Zhang BB, et al. Cell Metab 2009; 9:407 - 416
- Buzzai M, et al. Oncogene 2005; 24:4165 - 4173
- Yuneva M, et al. J Cell Biol 2007; 178:93 - 105
- Yang C, et al. Cancer Res 2009; 69:7986 - 7993
- Choo AY, et al. Mol Cell 38:487 - 499
- Yun J, et al. Science 2009; 325:1555 - 1559
- Chen V, et al. Cell Cycle 2010; 9:4941 - 4953
- Ito M, et al. Cancer Res 2003; 63:8763 - 8767
- Shtivelman E. Oncogene 1997; 14:2167 - 2173
Emerging evidence implicates that the TNF superfamily member B lymphocyte stimulator (BLyS), also known as (B-cell activating factor) BAFF, as well as its receptors, particularly BAFF-R, as critical factors for the growth and survival of both normal and malignant B cells.Citation1,Citation2 Although the signaling pathways mediated through BAFF-R have been extensively studied recently,Citation3–Citation5 the transcriptional regulation of BAFF-R has not been elucidated, especially in lymphomas and leukemias.
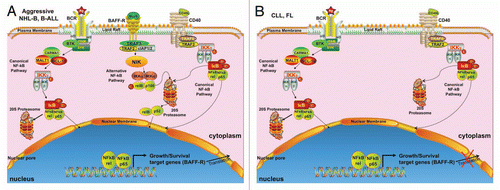
In a previous issue of Cell Cycle, Mihalcik and colleagues have shown that the TNF-receptor BAFF-R is dysregulated in two common human B-cell lymphoid lineage neoplasms with similar leukemic clinical presentations: precursor B-cell acute lymphoblastic leukemia (B-ALL) and Chronic Lymphocytic Leukemia (CLL). They first demonstrated that BAFF-R is constitutively expressed in early B-cell lines Reh and NALM-6, derived from patients with precursor B-ALL, and that BAFF-R is directly regulated by the canonical NF-kB member c-rel. Based on these findings, they suggested that BAFF-R plays a key role in growth and survival mechanisms of B-ALL. These findings are quite interesting because BAFF-R and c-rel are dysregulated in other types of aggressive NHL-B, such as diffuse large B-cell lymphomas, where c-rel interacts with CD40, another TNF receptor, regulating growth and survival genes.Citation4–Citation6 TNF-receptors like CD40 and BAFF-R can also enter the nucleus, interact with other transcription factors, and regulate gene transcription.Citation5–Citation7 BAFF-R/c-rel dysregulation in B-ALL could form a positive feedback mechanism, maintaining growth and survival of B-ALL, similar to other aggressive NHL-B cells (see ).
The authors further demonstrated that BAFF-R is down-regulated in CLL cells when compared to normal B cells, consistent with two previous findings by Briones et al.Citation8 and Lin et al.Citation9 Perhaps this is the most intriguing finding, since they showed that the transcriptional mechanism for BAFF-R expression in CLL is still intact, but the protein expression of BAFF-R and the binding affinity of BAFF to BAFF-R is absent/low. Post-transcriptional dysregulatory mechanism(s) are proposed but remain to be elucidated. Clearly, further studies are warranted to determine the biological significance as to why indolent lymphomas like follicular lymphoma (FL) and CLL cells down-regulate BAFF-R.
This report by Mihalcik et al. provides additional interesting studies in an area of particular recent interest and importance: the role of BLyS/BAFF-R, TNF ligand/receptor family that has been shown by them and others to be of central critical importance in key growth/survival mechanisms in both normal and neoplastic B cells. Minor caveats however, involve the selected neoplastic B-cell populations (e.g. very old B-ALL cell lines) studied that unfortunately but do not include patient B-ALL samples, or definitive clinical/phenotypic descriptions of the CLL patient samples that are PBMC populations, that are very heterogeneous in general, and may not ideal for studying the disease process. Better cellular choices would allow for a more valid assessment of how representative the studied cells are for defining the two disease entities. That being said, this study offers some insights in role of BAFF-R in the pathophysiology of two important B-cell malignancies. Although the mechanisms of deregulation of BAFF-R expression in B-cell malignancies remains unclear, understanding BAFF-R deregulation in these important cancers should offer novel therapeutic opportunities.
Figures and Tables
Figure 1 Schematic model for deregulation of BAFF-R expression in aggressive NHL-B, B-ALL, and indolent CLL and FL. (A) Aggressive lymphomas and leukemias, such DLBCL and B-ALL, respectively, constitutively express TNF-Receptors like CD40 and BAFF-R, as well as the B-cell receptor for mediating autonomous cell growth and survival mechanisms through internal deregulation of the ligand/receptor dyad, leading to the activation of the alternative and canonical NF-kB pathways. (B) Unlike aggressive lymphomas and leukemias, indolent lymphomas and leukemias such as CLL and FL, likely acquire key growth and survival signals from the outside environment. In addition, BAFF-R is down-regulated, possibly involving post-transcriptional mechanisms.
References
- Shivakumar L, et al. Clin Lymphoma Myeloma 2006; 7:106 - 108
- Woodland RT, et al. Semin Immunol 2006; 18:318 - 326
- Woodland RT, et al. Blood 2008; 111:750 - 760
- Pham LV, et al. Blood 2010; 116:3899 - 3906
- Fu L, et al. Blood 2009; 113:4627 - 4636
- Zhou HJ, et al. Blood 2007; 110:2121 - 217
- Lin-Lee YC, et al. J Biol Chem 2006; 281:18878 - 18887
- Briones J, et al. Exp Hematol 2002; 30:135 - 141
- Lin WY, et al. Blood 2007; 110:3959 - 3967