Abstract
Increasing chronological age is the most significant risk factor for cancer. Recently, we proposed a new paradigm for understanding the role of the aging and the tumor microenvironment in cancer onset. In this model, cancer cells induce oxidative stress in adjacent stromal fibroblasts. This, in turn, causes several changes in the phenotype of the fibroblast including mitochondrial dysfunction, hydrogen peroxide production, and aerobic glycolysis, resulting in high levels of L-lactate production. L-lactate is then transferred from these glycolytic fibroblasts to adjacent epithelial cancer cells and used as “fuel” for oxidative mitochondrial metabolism. Here, we created a new pre-clinical model system to directly test this hypothesis experimentally. To synthetically generate glycolytic fibroblasts, we genetically-induced mitochondrial dysfunction by knocking down TFAM using an sh-RNA approach. TFAM is mitochondrial transcription factor A, which is important in functionally maintaining the mitochondrial respiratory chain. Interestingly, TFAM-deficient fibroblasts showed evidence of mitochondrial dysfunction and oxidative stress, with the loss of certain mitochondrial respiratory chain components, and the over-production of hydrogen peroxide and L-lactate. Thus, TFAM-deficient fibroblasts underwent metabolic reprogramming towards aerobic glycolysis. Most importantly, TFAM-deficient fibroblasts significantly promoted tumor growth, as assayed using a human breast cancer (MDA-MB-231) xenograft model. These increases in glycolytic fibroblast driven tumor growth were independent of tumor angiogenesis. Mechanistically, TFAM-deficient fibroblasts increased the mitochondrial activity of adjacent epithelial cancer cells in a co-culture system, as seen using MitoTracker. Finally, TFAM-deficient fibroblasts also showed a loss of caveolin-1 (Cav-1), a known breast cancer stromal biomarker. Loss of stromal fibroblast Cav-1 is associated with early tumor recurrence, metastasis, and treatment failure, resulting in poor clinical outcome in breast cancer patients. Thus, this new experimental model system, employing glycolytic fibroblasts, may be highly clinically relevant. These studies also have implications for understanding the role of hydrogen peroxide production in oxidative damage and “host cell aging,” in providing a permissive metabolic microenvironment for promoting and sustaining tumor growth.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660) and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Figures and Tables
Figure 1 TFAM-deficient fibroblasts show a loss of Caveolin-1 protein expression. We generated TFAM-deficient immortalized fibroblast cell lines (hTERT-BJ1), using an sh-RNA approach. Note the successful knock-down of TFAM in stromal fibroblasts (sh-TFAM), as compared to control fibroblasts (sh-Ctrl), as seen by immuno-blot analysis. We also examined the status of the caveolin-1 (Cav-1) protein, because a loss of Cav-1 expression in the tumor stroma is a biomarker for tumor progression. Immuno-blot analysis shows that downregulation of TFAM also results in the loss of Cav-1 protein expression. Blotting for β-actin was performed in parallel as a control for equal protein loading.
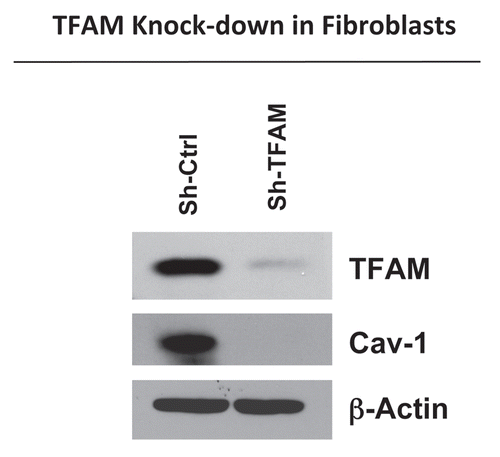
Figure 2 TFAM-deficient fibroblasts show defects in oxidative phosphorylation. To assess the status of the mitochondrial respiratory chain in fibroblasts, a battery of antibodies directed against mitochondrial complex components (I–V) were examined. Under normoxic conditions, no changes were observed between sh-TFAM and sh-Ctrl fibroblasts. However, under hypoxic conditions, note the decrease in Complexes I–IV. Immunoblotting with β-actin is shown as a control for equal loading.
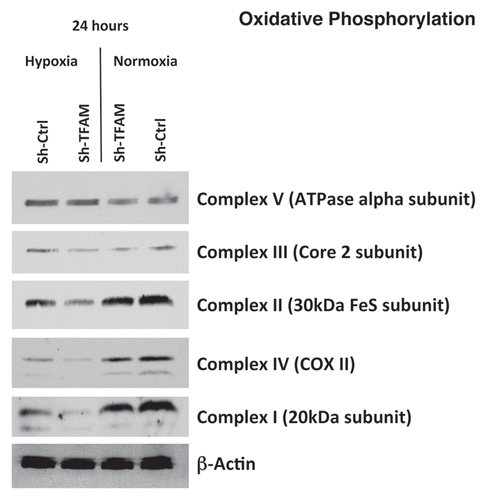
Figure 3 TFAM-deficient fibroblasts show a decrease in functional mitochondrial activity. We examined functional mitochondrial activity under normoxic conditions and found that sh-TFAM fibroblasts show a small, but significant, reduction (∼12%; p = 0.005) in activity.
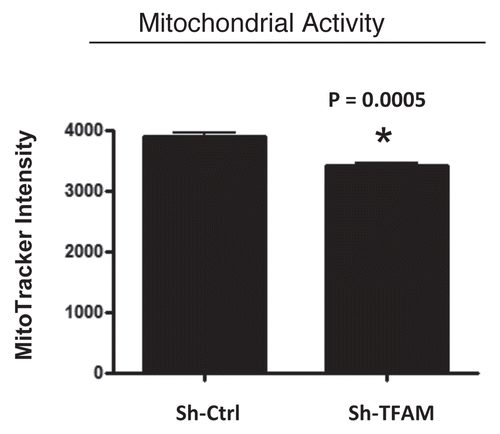
Figure 4 TFAM-deficient fibroblasts produce more hydrogen peroxide. Hydrogen peroxide production is significantly increased (∼2-fold; p = 0.001) in sh-TFAM fibroblasts, as compared to sh-Ctrl fibroblasts. The ability of TFAM-deficient fibroblasts to produce excess hydrogen peroxide suggests that a loss of TFAM expression could mimic the phenotypic behavior of tumor-associated myo-fibrobasts undergoing oxidative stress.
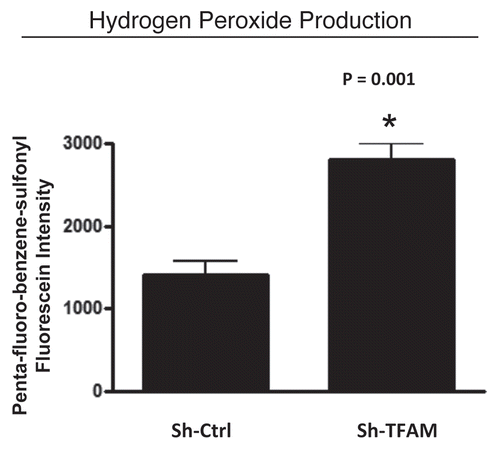
Figure 5 TFAM-deficient fibroblasts secrete elevated levels of L-lactate, consistent with increased aerobic glycolysis. L-lactate is a critical fuel which provides continued energetic support for tumor epithelial cells. Note that TFAM-deficient fibroblast secrete increased levels of L-lactate (∼2-fold; p = 0.006), relative to control fibroblasts processed in parallel.
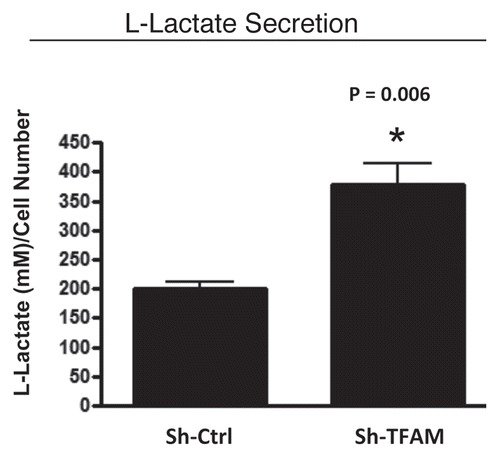
Figure 6 TFAM-deficient fibroblasts promote tumor growth. We used an in vivo murine xenograft model to evaluate the tumor-promoting properties of TFAM-deficient fibroblasts. Fibroblasts were co-injected with MDA-MB-231 breast cancer cells into the flanks of immuno-deficient nude mice. After 4 weeks, the tumors were harvested and subjected to detailed analysis. sh-TFAM fibroblasts were able to promote tumorigenesis, with an up to 2-fold increase in tumor growth. This represents an ∼1.6-fold increase (p = 0.03) in tumor weight and an ∼2.1-fold increase (p = 0.006) in tumor volume.
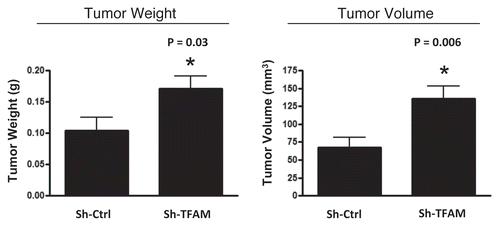
Figure 7 TFAM-deficient fibroblasts do not increase tumor angiogenesis. We used an in vivo murine xenograft model to evaluate the tumor-promoting properties of TFAM-deficient fibroblasts. Fibroblasts were co-injected with MDA-MB-231 breast cancer cells into the flanks of immuno-deficient nude mice. After 4 weeks, the tumors were harvested and subjected to detailed analysis. There were no observed differences in tumor vessel density density (number of vessels per field), as measured using CD31 immuno-staining. As such, the tumor growth promoting activity of TFAM-deficient fibroblasts is independent of tumor angiogenesis.
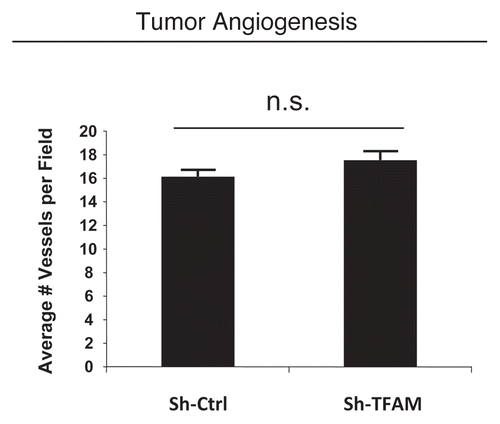
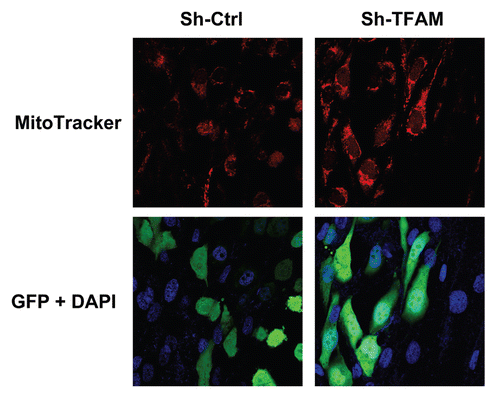
Figure 8 TFAM-deficient fibroblasts increase mitochondrial activity in adjacent cancer epithelial cells. We co-cultured sh-TFAM and sh-Ctrl fibroblasts with GFP-tagged MDA-MB-231 breast cancer cells. Then, we visualized mitochondrial activity in these co-cultures using MitoTracker. Note that TFAM-fibroblasts specifically increase the mitochondrial activity of adjacent MDA-MB-231 cells. MitoTracker (red); epithelial cancer cells/GFP (green); nuclei/DAP I (blue). See online version for color version of the figure.