Abstract
Colorectal cancer is a heterogeneous disease resulting from a combination of genetic and environmental factors. The C57BL/6J (B6) ApcMin/+ mouse develops polyps throughout the gastrointestinal tract and has been a valuable model for understanding the genetic basis of intestinal tumorigenesis. ApcMin/+ mice have been used to study known oncogenes and tumor suppressor genes on a controlled genetic background. These studies often utilize congenic knockout alleles, which can carry an unknown amount of residual donor DNA. The ApcMin model has also been used to identify modifer loci, known as Modifier of Min (Mom) loci, which alter ApcMin-mediated intestinal tumorigenesis. B6 mice carrying a knockout allele generated in WW6 embryonic stem cells were crossed to B6 ApcMin/+ mice to determine the effect on polyp multiplicity. The newly generated colony developed significantly more intestinal polyps than ApcMin/+ controls. Polyp multiplicity did not correlate with inheritance of the knockout allele, suggesting the presence of one or more modifier loci segregating in the colony. Genotyping of simple sequence length polymorphism (SSLP) markers revealed residual 129X1/SvJ genomic DNA within the congenic region of the parental knockout line. An analysis of polyp multiplicity data and SSLP genotyping indicated the presence of two Mom loci in the colony: 1) Mom12, a dominant modifier linked to the congenic region on chromosome 6, and 2) Mom13, which is unlinked to the congenic region and whose effect is masked by Mom12. The identification of Mom12 and Mom13 demonstrates the potential problems resulting from residual heterozygosity present in congenic lines.
Introduction
Colorectal cancer is one of the most common cancers in the United States, accounting for ∼10% of both cancer diagnoses and mortalities (www.cancer.org). The adenomatous polyposis coli (APC) gene is a tumor suppressor gene that is frequently mutated in both hereditary and sporadic colorectal cancers.Citation1,Citation2 APC encodes a large, multidomain protein that forms a complex with multiple other proteins, including AXIN and glycogen synthase kinase 3 beta (GSK3β).Citation3,Citation4 Formation of this complex results in negative regulation of WNT signaling through ubiquitination and degradation of the transcriptional co-activator β-catenin.Citation5–Citation8 Loss of functional APC results in nuclear accumulation of β-catenin, constitutive activation of cell growth via TCF/LEF transcription factors and tumor formation.Citation9–Citation11 Disease severity can vary in patients carrying identical APC mutations, indicating a role for genetic background in colorectal tumorigenesis (reviewed in ref. Citation12). The prevalence of colorectal cancer highlights the need for understanding the genetic factors underlying cancer development in the gastrointestinal tract.
Mouse models of colorectal cancer have been invaluable tools for identifying genetic and biochemical pathways that affect tumor initiation, growth and progression in the intestine.Citation13–Citation15 The multiple intestinal neoplasia (ApcMin/+) mouse carries a dominant mutation in the murine homolog of APC, which results in a premature stop codon and a truncated protein product.Citation16 The location of the mutation is analogous to mutations found in patients with familial adenomatous polyposis (FAP), a hereditary disorder resulting in the development of thousands of colon polyps.Citation17,Citation18 ApcMin/+ mice develop polyps in both the small intestine and colon following loss of the wild-type Apc allele through LOH and/or somatic mutation.Citation19,Citation20
Test crosses can be used to determine the effects of known mutations on ApcMin-mediated tumorigenesis. This reverse genetics approach allows prospective oncogenes and tumor suppressor genes to be studied in a system that recreates the genetic events in human colorectal cancer. Mutations in more than 50 genes have been analyzed on the ApcMin/+ background.Citation13,Citation21 In addition to testing specific genes, a forward genetics approach can identify novel modifier of Min (Mom) loci by using the genetic variation between C57BL/6J (B6) ApcMin/+ mice and other inbred strains.Citation22,Citation23 The ApcMin/+ mutation is maintained as a congenic line on a B6 genetic background. Crosses between B6 ApcMin>/+ and other inbred strains have demonstrated the presence of loci that affect phenotypes such as polyp multiplicity, size, location and stage (reviewed in ref. Citation13). Quantitative trait loci (QTL) analysis has been used to identify these modifier loci by correlating the inheritance of genetic markers across the genome with quantitative phenotypes.Citation21,Citation24 Six Mom loci have been described, with genes identified for two of the loci.Citation22,Citation23,Citation25–Citation28 By combining both forward and reverse genetics approaches, the ApcMin model has been an important tool for identifying and analyzing genes relevant to human colorectal cancer.
Caveolin-1 (CAV1) encodes an integral membrane protein, which oligomerizes to form microdomains known as caveolae.Citation29 Caveolae function as regulators of multiple signaling pathways, including pathways linked to cellular transformation, and the CAV1 gene has previously been identified as a tumor suppressor gene in human breast cancer.Citation29–Citation31 The CAV1 gene has also been linked to human colorectal cancer, but the role of the gene as either an oncogene or tumor suppressor gene is unclear.Citation32,Citation33 Loss of Cav1 expression in mice results in hyperplasia of stem cells in the intestinal crypt and the Cav1 protein sequesters beta-catenin and negatively regulates Wnt signaling.Citation34,Citation35 These data suggest that CAV1 may act as a tumor suppressor gene in the intestinal tract.
In this study, we sought to analyze the effect of loss of Cav1 gene expression on ApcMin-mediated tumorigenesis. Increased polyp multiplicity was observed in offspring of a B6 Cav1−/−x B6 ApcMin/+ cross compared to B6 ApcMin/+ controls. The genotype at the Cav1 locus, however, did not correlate with polyp multiplicity, implying the presence of other modifier loci segregating within the colony. Using simple sequence length polymorphism (SSLP) markers, we identified two novel modifier loci (Mom12 and Mom13) that affect intestinal tumorigenesis. Our data indicate that the susceptible Mom12 allele is linked to donor embryonic stem cell DNA flanking the Cav1 knockout allele on chromosome 6. The identification of the Mom12 and Mom13 loci highlights the potential effects of residual donor DNA on phenotypic analyses of congenic lines.
Results
Mice in a B6 Cav1−/− x B6 ApcMin/+ colony have increased intestinal tumorigenesis.
To observe the effects of loss of Caveolin-1 (Cav1) on intestinal tumorigenesis, female B6 Cav1−/− mice were mated to male B6 ApcMin/+ mice. The F1 offspring were back-crossed to the Cav1−/− parental strain. N2 offspring were then intercrossed to create a colony of mice carrying the ApcMin mutation in combination with all three potential Cav1 genotypes (Cav1−/−, Cav1−/+ and Cav1+/+). This colony is hereafter referred to as the “Cav1−/− x ApcMin/+ colony.” To determine if any modifier loci were segregating in the newly generated colony, we compared the polyp multiplicity of mice from the Cav1−/− x ApcMin/+ colony to ApcMin/+ control mice. At 120 days of age, mice from the Cav1−/− x ApcMin/+ colony have an average of 131 ± 50 polyps throughout the length of the intestinal tract (). The polyp multiplicity of mice from the B6 Cav1−/− x B6 ApcMin/+ colony is significantly higher than the polyp multiplicity of B6 ApcMin/+ controls (70 ± 33 polyps, p < 0.001). These data suggest that at least one modifier locus is segregating in the Cav1−/− x ApcMin/+ colony ().
Polyp density in B6 ApcMin/+ mice varies in different sections of the intestinal tract (). Different segments of the small intestine and colon are also known to have different functions and transcriptome profiles, suggesting a potential genetic basis for the variation in polyp multiplicity.Citation36 A modifier gene may, therefore, only affect tumorigenesis in one particular area of the intestine. To determine if the observed increase in polyp number in the Cav1−/− x ApcMin/+ colony is localized to a specific part of the intestinal tract, polyp multiplicities in four intestinal segments (proximal SI, middle SI, distal SI and colon) were compared between mice from the Cav1−/− x ApcMin/+ colony and ApcMin/+ controls. Mice from the Cav1−/− x ApcMin/+ colony had significantly more polyps in all areas of the small intestine than the ApcMin/+ controls (). Mice from the Cav1−/− x ApcMin/+ colony also have a higher colon polyp incidence (87%) than ApcMin/+ mice (67%, p < 0.001). These data indicate that the unknown modifier gene(s) in the colony affect in all areas of the intestinal tract.
Genotype at the Cav1 locus does not correlate with polyp multiplicity.
Since both the ApcMin/+ and Cav1−/− parental lines are maintained on a B6 genetic background, the most likely source of the increased polyposis in the Cav1−/− x ApcMin/+ colony is the Cav1 knockout allele. To confirm that the Cav1 knockout allele was functioning as a null allele, a western blot analysis was performed on tissue lysates from the distal small intestines of Cav1−/− mice. Transgenic B6 mice expressing myc-tagged Cav1 protein were used as controls for both normal Cav1 expression and antibody specificity. As expected, Cav1 was expressed in the control line but no expression of Cav1 protein was detected in Cav1−/− mice ().
After confirming the efficacy of the Cav1 knockout allele, we analyzed the effect of loss of Cav1 expression on ApcMin-mediated intestinal tumorigenesis. The Cav1 knockout allele may be acting as either a dominant or a recessive modifier gene. If the mutation is dominant, then both Cav1+/− and Cav1−/− mice will have increased polyp multiplicity. As a recessive allele, however, complete loss of Cav1 expression would be necessary for increased polyp multiplicity. Alternately, the Cav1 null allele may act as a semidominant modifier, with Cav1−/−, ApcMin/+ individuals having a more severe phenotype than Cav1+/−, ApcMin/+ individuals. The average polyp multiplicity of Cav1+/+, ApcMin/+ mice (143 ± 52 polyps), however, was not significantly different from the polyp multiplicities of either Cav1+/−, ApcMin/+ (125 ± 47 polyps, p = 0.13) or Cav1−/−, ApcMin/+ (123 ± 51 polyps, p = 0.11) mice (). The statistical analysis indicates that the increased tumorigenesis observed in the Cav1−/− x ApcMin/+ colony is not the result of decreased Cav1 expression.
Female mice from the Cav1−/− x ApcMin/+ colony develop more polyps than male mice.
To identify gender differences in tumorigenesis, polyp multiplicities in male and female mice were compared. Both male and female mice from the Cav1−/− x ApcMin/+ colony develop significantly more polyps than ApcMin/+ controls, indicating the modifiers in the colony affect polyposis in mice of both sexes (). Additionally, female mice from the Cav1−/− x ApcMin/+ colony develop significantly more polyps (144 ± 52) than male mice from the Cav1−/− x ApcMin/+ colony (122 ± 48, p < 0.03) (). The observed increase in polyp multiplicity in female mice could be specific to the colony and the result of a sex-specific modifier effect. Alternatively, the sex-specific differences in polyp multiplicity could be an intrinsic feature of the ApcMin/+ phenotype. To determine if the increased polyp multiplicity is unique to females in the Cav1−/− x ApcMin/+ colony, polyp multiplicities in male and female B6 ApcMin/+ mice were compared. Female B6 ApcMin/+ controls (78 ± 33 polyps) did not have significantly more polyps than male B6 ApcMin/+ controls (67 ± 33 polyps), although the difference was approaching significance (p = 0.1) (). Other laboratories have also reported an increase in polyp numbers in B6 ApcMin/+ females compared to B6 ApcMin/+ males, suggesting that the elevated polyp multiplicity in female mice is not specific to the Cav1−/− x ApcMin/+ colony.Citation37 To minimize the confounding factor of sex differences in polyp multiplicity, we analyzed male and female mice separately.
129X1 genomic DNA is linked to the Cav1 knockout allele.
The Cav1−/− line was generated using the WW6 ES cell line, which is derived from a cross between three different inbred strains: 129X1/SvJ (129X1), B6 and SJL.Citation38 Mice carrying the Cav1 knockout allele were then crossed to B6 mice for eight generations. At N8, ∼25 cM of genomic DNA linked to Cav1 is theoretically derived from the donor WW6 ES cell line.Citation38 If the WW6 cell line contains 129X1 or SJL genomic DNA around the Cav1 locus on proximal chromosome 6, the Cav1−/− mice may retain some of this non-B6 DNA. To determine if 129X1 genomic DNA is present in the Cav1−/− line, mice from the Cav1−/− x ApcMin/+ colony were genotyped for SSLP markers across the length of chromosome 6 (). 129X1 genomic DNA was observed at four markers distal to the Cav1 locus: D6Mit205, D6Mit268, D6Mit207 and D6Mit33. Only B6 DNA was found at marker D6Mit138, proximal to Cav1 and marker D6Mit316, distal to D6Mit33. These genotyping data define a 51 Mb congenic region on chromosome 6 that carries residual 129X1 genomic DNA ().
Two modifier loci are segregating in the Cav1−/− x ApcMin/+ colony.
As the discovery of Mom1 previously demonstrated, polymorphisms between inbred strains can modify the polyposis phenotype observed in mice carrying the ApcMin mutation (reviewed in ref. Citation13). A polymorphism between 129X1 and B6 near the Cav1 locus may, therefore, result in the increased tumorigenesis observed in the Cav1−/− x ApcMin/+ colony. To determine if a modifier gene is present in the congenic region, we examined the distribution of the polyp multiplicity data in the Cav1−/− x ApcMin/+ colony. A bimodal distribution may indicate the segregation of a single dominant modifier allele, while three phenotypic groups may indicate a semidominant mode of inheritance. To explore the relationship between genotype and polyp multiplicity, the distribution of polyp multiplicity data was analyzed based on the genotypes at five loci within the maximum congenic region. No correlation was found for the Cav1, D6Mit205, D6Mit268 or D6Mit207 loci (data not shown). The D6Mit33 marker, however, correlated with the distribution of polyp multiplicity. Both male and female mice with the B6/B6 genotype at the D6Mit33 marker had an apparent bimodal distribution ( and B). The high polyp multiplicity group from each sex (female: >150 polyps, male: >110 polyps) had significantly more polyps than ApcMin/+ controls (female: p < 0.001, male: p < 0.001). Mice with the 129/B6 genotype at the D6Mit33 marker had a distribution with a single peak centered between the two B6/B6 groups ( and D). The single 129/B6 group was significantly different than the low and high B6/B6 groups in both male and female mice (female: p < 0.01, male: p < 0.05). The polyp multiplicity of 129/B6 mice (female: 150 ± 47 polyps, male: 130 ± 37 polyps) was also higher than the polyp multiplicity of ApcMin/+ controls (female: 78 ± 33 polyps, p < 0.001, male: 67 ± 33 polyps, p < 0.001).
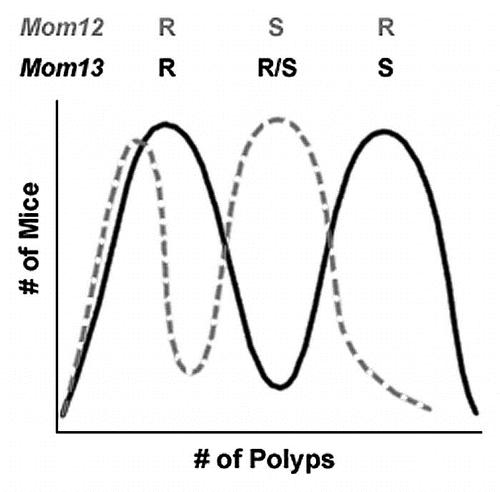
The most likely hypothesis explaining these three phenotypic groups is the existence of two dominant modifier loci segregating in the Cav1−/− x ApcMin/+ colony (). One dominant modifier, hereafter referred to as Mom12, is linked to the 129X1 allele of the D6Mit33 marker on chromosome 6. The presence of the Mom12 susceptible allele results in an increase in polyp multiplicity compared to ApcMin/+ controls ( and D). A second dominant modifier, hereafter referred to as Mom13, is unlinked to the congenic region. In the absence of the Mom12 susceptible allele, the Mom13 susceptible allele results in increased polyp multiplicity ( and B).
Discussion
The ApcMin/+ mouse is a commonly used model for determining the mechanism underlying intestinal tumorigenesis and identifying genes that are integral to tumor initiation and growth (reviewed in ref. Citation13). There is a large volume of evidence indicating a role for Cav1 as a negative regulator of both cancer-associated phenotypes, such as increased proliferation and disturbed cell cycle progression, as well as multiple signaling pathways including WNT signaling (reviewed in ref. Citation29). Negative regulation of the WNT pathway would suggest that Cav1 is a prospective tumor suppressor in colorectal cancer. Cav1−/− mice also have increased proliferation of intestinal stem cells, supporting the tumor suppressor gene hypothesis.Citation34 If Cav1 were acting as a tumor suppressor gene in the colon, expression of the protein would be expected to decrease in transformed colon cells. In one study, decreased expression was found in multiple colon cancer cell lines.Citation33 The exact opposite, however, was seen in two similar studies.Citation39,Citation40 It is possible that these conflicting results are due to the use of cell lines, which often acquire further mutations that are selected for by the culture environment. The same conflicting evidence, however, has been observed in primary colon tumor samples.Citation32,Citation33 The role of Cav1 in colon cancer currently remains unclear.
The present study was originally designed to determine the effect of Cav1 loss on intestinal tumorigenesis on an inbred genetic background. If Cav1 functions as a tumor suppressor gene in colorectal cancer, an increase in polyp multiplicity in Cav1−/− mice would be expected. A cross between B6 Cav1−/− and B6 ApcMin/+ mice resulted in a colony with a higher polyp multiplicity than B6 ApcMin/+ controls, suggesting the segregation of a modifier gene in the colony. Although the Cav1 knockout allele was shown to successfully eliminate expression of the Cav1 protein (), the genotype at the Cav1 locus was not found to correlate with polyp multiplicity ().
These data suggest that Cav1 does not act as a tumor suppressor gene in intestinal tumorigenesis. However, a recent study demonstrated that reintroduction of full length APC transcript into human colon tumor cells results in upregulation of CAV1.Citation41 The study suggests a mechanism by which CAV1 expression is regulated downstream of APC. Mutations in the APC gene are early genetic lesions in almost all cases of colorectal cancer. If CAV1 expression is controlled downstream of APC, the loss of APC in colorectal tumors may result in downregulation of CAV1. In this scenario, colon-specific phenotypes caused by the loss of the CAV1 gene may be a subset of the phenotypes observed following APC mutations. The addition of a Cav1 null allele might not alter the ApcMin phenotype because Cav1 expression is already downregulated in the tumor tissue following the loss of the wild-type Apc allele. Additionally, the presence of two modifier loci segregating in the Cav1−/− x ApcMin/+ colony could be masking any effects of Cav1. Similar interactions have been observed with the resistant allele of the Mom1 locus, which is potent enough to mask the presence of other modifier loci. Further studies of Cav1−/−, ApcMin/+ mice will have to be performed in the absence of the Mom12 and Mom13 susceptible alleles.
The elimination of Cav1 as the cause of the observed increase in polyp multiplicity indicates the presence of at least one unknown modifier still segregating in the colony. SSLP genotyping of chromosome 6 identified a 51 Mb region of 129X1 genomic DNA flanking the Cav1 knockout allele (); however, no correlation was found between genotype and polyp multiplicity for any of the SSLP markers on chromosome 6. Further analysis found a correlation between the genotype at the D6Mit33 marker and the distribution of polyp multiplicities. Mice with a B6/B6 genotype at D6Mit33 had an apparent bimodal distribution: one phenotypic group was not statistically different than ApcMin/+ mice, while the other group developed a larger number of polyps than ApcMin/+ mice. As demonstrated by the identification of Mom2 and Mom7, this bimodal pattern is a hallmark of a dominant modifier gene.Citation23,Citation25 The polyp multiplicity data for mice with the 129/B6 genotype at D6Mit33 formed a third phenotypic group that was statistically different than either the high or low groups observed in B6/B6 mice. This third group also has significantly more polyps than ApcMin/+ controls, indicating a second modifier is also present in the colony.
The polyp multiplicity data indicate the presence of two dominant modifier loci that are independently segregating in the Cav1−/− x ApcMin/+ colony (). One modifier, Mom12, is linked to the D6Mit33 marker on chromosome 6 and results in increased tumorigenesis compared to ApcMin/+ controls. A second modifier, Mom13, is unlinked to the Cav1 locus and increases polyp multiplicity in the absence of the Mom12 susceptible (Mom12S) allele. The Mom13 locus may be the result of either a spontaneous mutation arising in one of the parental lines or residual 129X1 DNA segregating in the parental Cav1−/− line. If Mom12 and Mom13 are either additive or synergistic, two phenotypic groups would be expected in mice with a 129/B6 genotype at the D6Mit33 marker. Only one group, however, is observed in mice with this genotype. The single phenotypic group could be explained by epistasis; Mom12 being epistatic to Mom13 in the Cav1−/− x ApcMin/+ colony would result in the Mom12 phenotype being present regardless of Mom13 genotype. The presence of one Mom12S allele alone results in a moderate increase in polyp multiplicity compared to B6 ApcMin/+ controls. The presence of one Mom13S allele results in a larger increase in polyp multiplicity. If both modifiers are present, however, the Mom12S allele masks the phenotype of Mom13 and only the moderate increase in polyp multiplicity associated with Mom12 is observed (). Alternately, the two modifier loci segregating in the colony may be mutually exclusive due to embryonic lethality. If mice carrying susceptible alleles from both modifier genes are not viable, then none of the mice with a 129/B6 genotype at the D6Mit33 marker could be carrying a Mom13S allele. This scenario would also explain the lack of a second phenotypic group in mice with the 129/B6 genotype at D6Mit33.
In addition to providing another modifier gene for identification and further study, the discovery of the Mom12 locus underscores potential problems with congenic mouse lines. Previous studies have shown that passenger genes within the congenic region can have unforeseen effects on the phenotype of interest, acting as confounding variables during an analysis.Citation42 Until recently, B6 ES cells were not commonly used for the generation of transgenic mouse lines due to difficulties deriving ES cell lines from the inbred strain.Citation43 129X1 ES cells were not only easier to isolate, but better able to colonize the germ-line in chimeric mice (reviewed in ref. Citation44). As a result, most ES cell lines used for creating transgenic mice are derived from the 129X1 inbred strain. B6 is one of the most commonly studied inbred backgrounds and both transgenic and gene-targeted mice are often made congenic on the B6 background. Backcrossing to the B6 inbred strain can, therefore, create a scenario in which the donor and recipient genomes of a congenic line are not derived from the same inbred strain. The discovery of the Mom12 locus underlies the potential problem with this methodology: it is impossible to completely remove donor DNA from a transgenic mouse line. A transgenic mouse line will always carry a region of ES cell genomic DNA linked to the transgenic allele. Numerous studies have demonstrated that polymorphisms between inbred strains can affect quantitative traits (reviewed in ref. Citation13). If the donor and recipient strains of a transgenic line are different, then residual ES cell DNA may affect the phenotype being studied. In the case of Mom12, the modifier effect was distinguishable from the effects of Cav1 because the D6Mit33 marker is 33.5 Mb distal to the Cav1 locus. However, the closer a modifier gene is to the transgenic allele, the higher the probability that the effects of the two loci would be indistinguishable. The phenotype caused by an unknown modifier locus may, therefore, be falsely attributed to the gene of interest.
Materials and Methods
Generation of the Cav1−/− x ApcMin/+ colony.
Production of the Cav1 knockout mouse line has been previously described in reference Citation45. Briefly, a targeting construct carrying a neomycin resistance cassette was inserted into the pGT-N29 targeting vector. The NEO cassette was flanked by the genomic sequences immediately upstream of exon 1 and immediately downstream of exon 2 of the Cav1 gene. The targeting vector was electroporated into WW6 embryonic stem cells. ES cells were selected for the presence of the NEO cassette and microinjected into C57BL/6J (B6) blastocysts to generate chimeric offspring. Genomic DNA was isolated from tail biopsies using the DNeasy Kit (Qiagen, Cat. No. 69506) and mice were genotyped by PCR for the presence of the knockout allele (Wild Type Forward Primer: GTG TAT GAC GCG CAC ACC AAG, Knockout Forward Primer: CTA GTG AGA CGT GCT ACT TCC, Common Reverse Primer: CTT GAG TTG AGT TCT GTT AGC CCA G). Mice carrying the knockout allele were backcrossed to B6 mice from The Jackson Laboratory for eight generations and then intercrossed to generate a B6 Cav1−/− line. B6 Cav1−/− female mice were mated to B6 ApcMin/+ male mice from The Jackson Laboratory. F1 offspring were back-crossed to B6 Cav1−/− female mice. N2 offspring were intercrossed to generate a colony of mice carrying the ApcMin mutation with the Cav1+/+, Cav1−/+ or Cav1−/− genotypes. The colony was maintained in the AALAC-accredited TJU animal facility.
Evaluation of intestinal tumorigenesis in the Cav1−/− x ApcMin/+ colony.
Mice at 120 days of age were euthanized by CO2 asphyxiation followed by cervical dislocation. The intestinal tract was washed with 1x PBS and separated into four segments: proximal small intestine (SI), middle SI, distal SI and colon. Polyp multiplicity was determined for each segment with a Nikon SMZ-U dissection microscope at 22.5× magnification.
Western blot analysis.
B6 Cav1−/− mice, B6 Cav1Tg mice and B6 controls at 60 days of age were euthanized by CO2 asphyxiation followed by cervical dislocation. Distal small intestine samples were washed in 1x PBS and homogenized in lysis buffer (50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 2 mM EDTA; 10% glycerol; 1% Triton-X-100) with protease inhibitors (Roche Applied Science, Cat. No. 11836153001). Protein concentration was quantified using Bio-Rad Protein Assay (Bio-Rad Laboratories, Cat. No. 500-0006). 20 µg of each sample was separated by SDS-PAGE (10% acrylamide) and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in 1x TBST (20 mM Tris-HCl, pH 7.4; 150 mM NaCl; 0.05% Tween 20). Anti-caveolin-1 antibody N-20 (Santa Cruz Biotechnology, Cat. No. SC-894) was used at a 1:1,000 dilution overnight at 4°C. Horseradish peroxidase-conjugated secondary antibody (Vector Laboratories, Cat. No. PI-1000) was used at a 1:2,500 dilution for 1 h at room temperature. Proteins were visualized using SuperSignal chemiluminescent substrate (Thermo Scientific, Cat. No. 34080).
Simple sequence length polymorphism (SSLP) genotyping.
Polymorphic markers between the 129X1/SvJ (129X1) and B6 inbred strains were identified using the Mouse Microsatellite Database of Japan (www.shigen.nig.ac.jp/mouse/mmdbj). Only markers with at least a 4 bp difference between the 129X1 and B6 alleles were chosen to ensure adequate resolution of the two alleles. Primer sequences were obtained from the Ensembl Genome Browser (www.ensembl.org). Genomic DNA was isolated from tail biopsies by DNeasy Kit (Qiagen, Cat. No. 69506). SSLP markers were amplified from ∼50 ng of genomic DNA using Choice Taq Blue DNA Polymerase (Denville Scientific, Cat. No. CB4060-3) under the following conditions: one cycle at 94°C for 5 min, followed by 35 cycles of 94°C for 30 sec, 57°C for 30 sec, 72°C for 30 s and ending with 72°C for 7 min. Polymorphisms were detected on 4% TBE agarose gels and visualized with ethidium bromide.
Abbreviations
| 129X1 | = | 129X1/SvJ |
| APC | = | adenomatous polyposis coli |
| B6 | = | C57BL/6J |
| CAV1 | = | caveolin-1 |
| Mom | = | modifier of min |
| SSLP | = | simple sequence length polymorphism |
Figures and Tables
Figure 1 Comparison of polyp number and location between ApcMin/+ mice and mice from the Cav1−/−x ApcMin/+ colony at 120 days of age. (A) Average polyp number detected in the small intestine and colon combined. (B) Average polyp numbers detected in the proximal, middle and distal portion of the small intestine and colon. (*p < 0.05. **p < 0.005. ***p < 0.001. n = 147 for ApcMin/+ mice and n = 104 for mice from the Cav1−/−x ApcMin/+ colony. Error bars represent standard deviation).
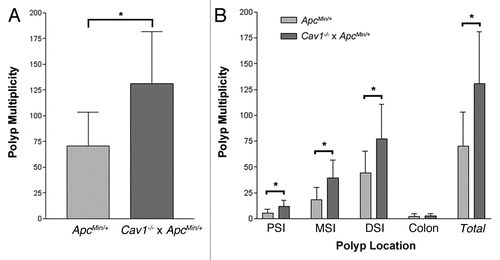
Figure 2 Cav1 genotype does not correlate with intestinal polyp number in ApcMin/+ mice. (A) Distal small intestine samples were taken from B6 Cav1−/− mice and transgenic mice expressing Myc-tagged Cav1 (B6 Cav1Tg). Cav1 expression was analyzed by western blot. Actin was used as a protein loading control. (B) Average polyp numbers detected in the intestine and colon of ApcMin/+ mice with Cav1+/+, Cav1−/+ or Cav1−/− genotypes. (n = 39 for Cav1+/+ mice, n = 33 for Cav1−/+ mice and n = 32 for Cav1−/− mice. Error bars represent standard deviation).
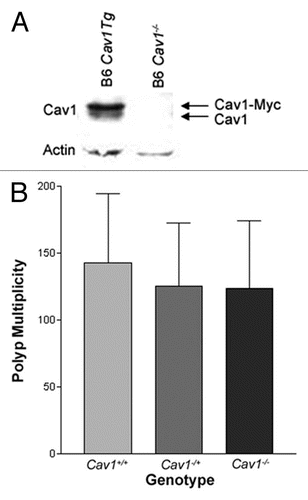
Figure 3 Female mice from the Cav1−/−x ApcMin/+ colony develop more polyps than male mice at 120 days of age. Average polyp number detected in the small intestine and colon combined. (*p < 0.05; **p < 0.005; ***p < 0.001; n = 46 for female mice and n = 58 for male mice from the Cav1−/−x ApcMin/+ colony; n = 50 for female ApcMin/+ mice and n = 97 for male ApcMin/+ mice. Error bars represent standard deviation).
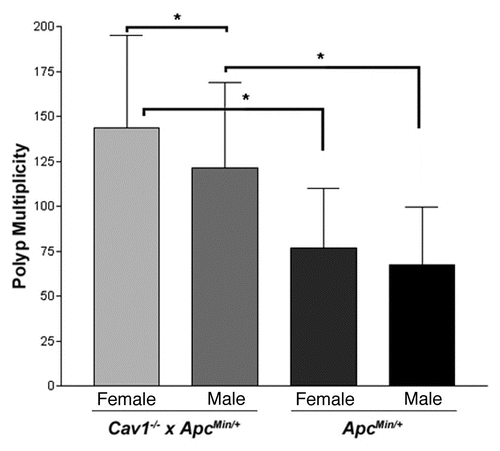
Figure 4 Map of residual 129X1/SvJ genomic DNA in the congenic region of the Cav1 knockout allele. Tail DNA was tested for SSLP markers across the length of chromosome 6. Marker locations are given in Mb from the centromere. Gray boxes indicate the presence of 129X1 genomic DNA. Black boxes indicate the absence of 129X1 genomic DNA. The maximum congenic region is a 51 Mb area between the D6Mit138 and D6Mit316 markers.
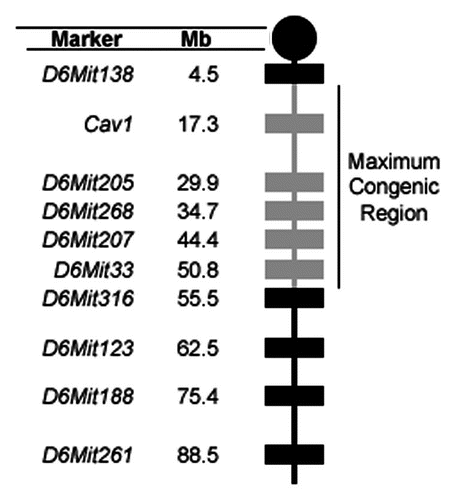
Figure 5 Polyp multiplicity for mice in the Cav1−/−x ApcMin/+ colony based on sex and D6Mit33 genotype. Polyp number data includes both small intestine and colon. Female mice were separated into (A) B6/B6 (n = 25) and (B) 129/B6 (n = 18) genotypes. High polyp multiplicity in B6/B6 female mice was defined as >150 polyps. Male mice were also separated into (C) B6/B6 (n = 35) and (D) 129/B6 (n = 14) genotypes. High polyp multiplicity in B6/B6 male mice was defined as >110 polyps.
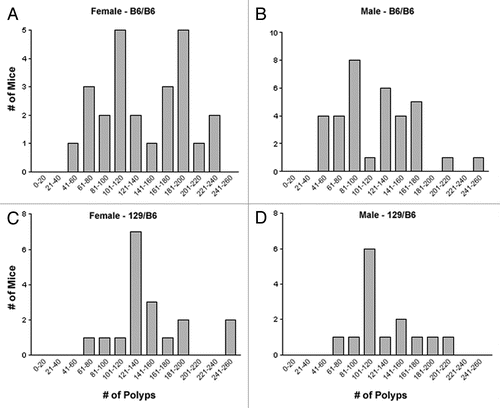
Figure 6 Representation of the effects of Mom12 and Mom13 on intestinal tumorigenesis. the effect of Mom12 on polyp number is represented by the dashed gray curve. the effect of Mom13 is represented by the black curve. Both modifiers individually have an apparent bimodal distribution. Mice with resistant alleles of both modifiers develop the same number of polyps as B6 ApcMin/+ controls. Mice carrying a susceptible allele at either Mom12 or Mom13 have increased polyp numbers. Mice carrying susceptible alleles at both modifier loci have the Mom12 phenotype. Expected genotypes for each modifier locus are listed above the phenotypic groups. (R, resistant allele; S, susceptible allele).
Acknowledgements
We thank Stephanie Nnadi for critical reading of the manuscript. This work was supported by the National Institutes of Health [T32-CA09678 to R.C.C., T32-CA09683 to J.J.R.]; and the National Cancer Institute.
References
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992; 359:235 - 237
- Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, et al. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res 1994; 54:3011 - 3020
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996; 272:1023 - 1026
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004; 20:781 - 810
- Pai LM, Orsulic S, Bejsovec A, Peifer M. Negative regulation of Armadillo, a Wingless effector in Drosophila. Development 1997; 124:2255 - 2266
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase3. Genes Dev 1996; 10:1443 - 1454
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 1997; 16:3797 - 3804
- Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem 1997; 272:24735 - 24738
- Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, et al. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 1999; 96:1603 - 1608
- Polakis P, Hart M, Rubinfeld B. Defects in the regulation of beta-catenin in colorectal cancer. Adv Exp Med Biol 1999; 470:23 - 32
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996; 86:391 - 399
- Fodde R, Smits R. Disease model: familial adenomatous polyposis. Trends Mol Med 2001; 7:369 - 373
- Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv Exp Med Biol 2009; 656:85 - 106
- Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology 2009; 136:780 - 798
- Uronis JM, Threadgill DW. Murine models of colorectal cancer. Mamm Genome 2009; 20:261 - 268
- Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990; 247:322 - 324
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66:589 - 600
- Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991; 253:665 - 669
- Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, et al. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet 2002; 11:1549 - 1560
- Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res 1994; 54:5947 - 5952
- Siracusa LD, Silverman KA, Koratkar R, Markova M, Buchberg AM. Brenner C, Duggan D. Genome-wide modifier screens: How the genetics of cancer penetrance may shape the future of prevention and treatment. Oncogenomics: Molecular Approaches to Cancer 2004; New Jersey John Wiley & Sons, Inc 255 - 290
- Gould KA, Dietrich WF, Borenstein N, Lander ES, Dove WF. Mom1 is a semi-dominant modifier of intestinal adenoma size and multiplicity in Min/+ mice. Genetics 1996; 144:1769 - 1776
- Kwong LN, Shedlovsky A, Biehl BS, Clipson L, Pasch CA, Dove WF. Identification of Mom7, a novel modifier of Apc(Min/+) on mouse chromosome 18. Genetics 2007; 176:1237 - 1244
- Huang H, Eversley CD, Threadgill DW, Zou F. Bayesian multiple quantitative trait loci mapping for complex traits using markers of the entire genome. Genetics 2007; 176:2529 - 2540
- Silverman KA, Koratkar R, Siracusa LD, Buchberg AM. Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Res 2002; 12:88 - 97
- Suraweera N, Haines J, McCart A, Rogers P, Latchford A, Coster M, et al. Genetic determinants modulate susceptibility to pregnancy-associated tumourigenesis in a recombinant line of Min mice. Hum Mol Genet 2006; 15:3429 - 3435
- Oikarinen SI, Cleveland AG, Cork KM, Bynote KK, Rafter JJ, Gustafsson JA, et al. Genetic mapping of Mom5, a novel modifier of Apc(Min)-induced intestinal tumorigenesis. Carcinogenesis 2009; 30:1591 - 1596
- Cormier RT, Bilger A, Lillich AJ, Halberg RB, Hong KH, Gould KA, et al. The Mom1AKR intestinal tumor resistance region consists of Pla2g2a and a locus distal to D4Mit64. Oncogene 2000; 19:3182 - 3192
- Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer and metastasis. Am J Physiol Cell Physiol 2005; 288:494 - 506
- Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE. Tumor cell growth inhibition by caveolin reexpression in human breast cancer cells. Oncogene 1998; 16:1391 - 1397
- Zou W, McDaneld L, Smith LM. Caveolin-1 haploinsufficiency leads to partial transformation of human breast epithelial cells. Anticancer Res 2003; 23:4581 - 4586
- Fine SW, Lisanti MP, Galbiati F, Li M. Elevated expression of caveolin-1 in adenocarcinoma of the colon. Am J Clin Pathol 2001; 115:719 - 724
- Bender FC, Reymond MA, Bron C, Quest AF. Caveolin-1 levels are downregulated in human colon tumors, and ectopic expression of caveolin-1 in colon carcinoma cell lines reduces cell tumorigenicity. Cancer Res 2000; 60:5870 - 5878
- Li J, Hassan GS, Williams TM, Minetti C, Pestell RG, Tanowitz HB, et al. Loss of caveolin-1 causes the hyper-proliferation of intestinal crypt stem cells, with increased sensitivity to whole body gamma-radiation. Cell Cycle 2005; 4:1817 - 1825
- Galbiati F, Volonte D, Brown AM, Weinstein DE, Ben-Ze'ev A, Pestell RG, et al. Caveolin-1 expression inhibits Wnt/beta-catenin/Lef-1 signaling by recruiting beta-catenin to caveolae membrane domains. J Biol Chem 2000; 275:23368 - 23377
- Comelli EM, Lariani S, Zwahlen MC, Fotopoulos G, Holzwarth JA, Cherbut C, et al. Biomarkers of human gastrointestinal tract regions. Mamm Genome 2009; 20:516 - 527
- McAlpine CA, Barak Y, Matise I, Cormier RT. Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. Int J Cancer 2006; 119:2339 - 2346
- Ioffe E, Liu Y, Bhaumik M, Poirier F, Factor SM, Stanley P. WW6: an embryonic stem cell line with an inert genetic marker that can be traced in chimeras. Proc Natl Acad Sci USA 1995; 92:7357 - 7361
- Patlolla JM, Swamy MV, Raju J, Rao CV. Overexpression of caveolin-1 in experimental colon adenocarcinomas and human colon cancer cell lines. Oncol Rep 2004; 11:957 - 963
- Burgermeister E, Tencer L, Liscovitch M. Peroxisome proliferator-activated receptor-gamma upregulates caveolin-1 and caveolin-2 expression in human carcinoma cells. Oncogene 2003; 22:3888 - 3900
- Roy UK, Henkhaus RS, Ignatenko NA, Mora J, Fultz KE, Gerner EW. Wild-type APC regulates caveolin-1 expression in human colon adenocarcinoma cell lines via FOXO1a and C-myc. Mol Carcinog 2008; 47:947 - 955
- de Ledesma AM, Desai AN, Bolivar VJ, Symula DJ, Flaherty L. Two new behavioral QTLs, Emo4 and Reb1, map to mouse Chromosome 1: Congenic strains and candidate gene identification studies. Mamm Genome 2006; 17:111 - 118
- Seong E, Saunders TL, Stewart CL, Burmeister M. To knockout in 129 or in C57BL/6: that is the question. Trends Genet 2004; 20:59 - 62
- Ledermann B. Embryonic stem cells and gene targeting. Exp Physiol 2000; 85:603 - 613
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 2001; 276:38121 - 38138