Abstract
Comment on: Basu A, et al. PLoS One 2011; 6:e23919
The development and rapid progression of cancer is a major problem in immunosuppressed patients, including patients receiving organ transplants, and renal cancer is one of the most common cancers during post-transplantation period.Citation1 Although improvements in immunosuppressive therapies have markedly reduced the incidence of acute allograft rejection, they are also associated with higher rates of mortality due to an increased risk of cancer.Citation1 Interestingly, it has been observed that transplant patients receiving the mammalian target of rapamycin (mTOR) inhibitor rapamycin (RAPA) do not develop cancer at the rate as those receiving other immunosuppressive agents, such as calcineurin inhibitors (CNIs).Citation2 Classically, CNIs inhibit calcineurin to prevent the activation of NFAT.Citation3 However, apart from inhibiting NFAT, CNIs may also regulate other signaling molecules playing important roles in tumor growth. Previously, we defined a novel mechanism by which CNIs can activate the proto-oncogene ras and specific isoforms (ζ and δ) of protein kinase C (PKC);Citation4–Citation6 and CNIs can promote a rapid progression of human renal cancer through transcriptional activation of VEGF and VEGF-induced angiogenesis.Citation5 In contrast to CNIs, RAPA treatment may have an anti-angiogenic effect.Citation2
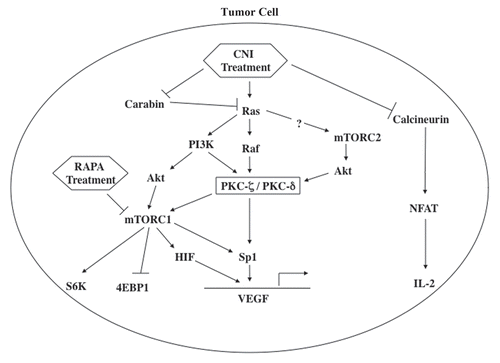
In our most recent study in reference Citation7, we observed that RAPA treatment can significantly downregulate CNI-induced and Ras-mediated overexpression of VEGF in renal cancer cells, suggesting involvement of the mTOR pathway in CNI-induced tumorigenic processes. To confirm this, we have shown that the knockdown of raptor significantly decreased CNI-induced VEGF promoter activity, indicating the role of mTOR complex1 (mTORC1),Citation8 in CNI-induced VEGF transcription. We found that the treatment with CNI cyclosporine (CsA) and activation of the Ras-PKC pathway can increase the phosphorylation of proline-rich Akt substrate of 40 kDa (PRAS40), a negative regulator of mTORC1.Citation8,Citation9 Thus, CNI-induced signals may relieve the inhibition of mTOR through the regulation of PRAS40. Our study identifies a novel crosstalk between CNI-induced signaling molecules (Ras and PKC) and mTOR for the regulation of VEGF expression in renal cancer cells ().Citation7
What can be the broader implications of our study? As mentioned earlier, CNIs (CsA and FK506) are excellent immunosuppressive agents to inhibit allograft rejection; however, they may promote the growth of tumors, including renal tumors in transplant patients.Citation1 Thus, it is a challenge for the clinicians to fix a safe but effective immunosuppressive agent for the treatment of transplant patients to achieve optimal immunosuppression as well as to prevent cancer development/recurrence. The mechanism(s) underlying the development/rapid progression of post-transplantation cancer should be thoroughly evaluated, such that specific agents can be used to target cancer development/progression. It is known that renal tumors are highly angiogenic, and they express VEGF.Citation5 Our data indicate that CNI treatment promotes the overexpression of VEGF,Citation5,Citation6 which can turn on the angiogenic switch in renal cancer cells. Thus, CNI-induced and VEGF-mediated angiogenesis may facilitate a rapid progression of dormant tumors in transplant patients. Interestingly, our recent observations suggest that the mTOR pathway plays a significant role in CNI-induced and VEGF-mediated tumorigenic processes.Citation7 Thus, a combination therapy using both CNI and RAPA may play a beneficial role in inhibiting allograft rejection as well as preventing cancer development in transplant patients. Of importance, mTOR is often activated in different types of cancer cells, including renal cancer. Mutations in Ras, PI3K, PTEN and VHL may lead to mTOR activation, and it is now established that treatment with RAPA can decrease cancer incidence in both animals and humans.Citation2,Citation10
Our data also show a novel crosstalk among mTOR, Ras and specific isoforms of PKC, particularly PKCζ and PKCδ, for the regulation of CNI-induced VEGF expression (), and both PKCζ and PKCδ formed complexes with PRAS40.Citation7 We found that the Ras-induced phosphorylation of PRAS40 was inhibited in the presence of PKC inhibitor. Thus, we suggest that specific inhibitors of PKC isoforms may significantly limit CNI-induced and VEGF-mediated renal tumor growth in transplant patients. However, our work did not define if there is a direct complex formation between PKC isoforms and PRAS40, and whether some other associated molecules are involved in Ras-PKC-mediated PRAS40 phosphorylation. In addition, the CNI-induced Ras activation may also be a complex process and needs further study. For example, it has been reported that a novel endogenous molecule carabin may inhibit not only the calcineurin pathway, but also the Ras signaling pathway,Citation11 and we previously observed that CNI treatment can downregulate carabin, and thereby may promote Ras activation.Citation4 Thus, identifying novel target molecules regulating the complex network of CNI-induced and Ras-PKC-mTOR-mediated pathway(s) for VEGF expression may open up new therapeutic avenues for the treatment of post-transplantation cancer.
Figures and Tables
Figure 1 Potential crosstalk among Ras, PKCζ/PKCδ and the mTOR signaling pathway(s) regulating calcineurin inhibitor (CNI)-induced VEGF overexpression in renal cancer cells. Following CNI treatment, the Ras-PKC pathway becomes activated and promotes transcriptional activation of VEGF through the transcription factor Sp1. Carabin may play a significant role in CNI-induced Ras activation. CNI-induced and Ras-PKC-mediated signals can also be channeled through mTOR for the regulation of VEGF expression; Ras- and PKC (PKCζ/PKCδ)-mediated signals can promote the activation mTOR complex1 (mTORC1). Together, mTOR may act as a critical intermediary signaling molecule for CNI-induced VEGF overexpression in renal cancer cells.
Acknowledgements
This work was supported by National Institutes of Health Grant R01 CA131145 (to S.P.).
Comment on: Basu A, et al. PLoS One 2011; 6:e23919
References
- Kasiske BL, et al. Am J Transplant 2004; 4:905 - 913; PMID: 15147424; http://dx.doi.org/10.1111/j.1600-6143.2004.00450.x
- Guba M, et al. Nat Med 2002; 8:128 - 135; PMID: 11821896; http://dx.doi.org/10.1038/nm0202-128
- Liu J, et al. Cell 1991; 66:807 - 815; PMID: 1715244; http://dx.doi.org/10.1016/0092-8674(91)90124-H
- Datta D, et al. Cancer Res 2009; 69:8902 - 8909; PMID: 19903851; http://dx.doi.org/10.1158/0008-5472.CAN-09-1404
- Basu A, et al. Cancer Res 2008; 68:5689 - 5698; PMID: 18632621; http://dx.doi.org/10.1158/0008-5472.CAN-07-6603
- Basu A, et al. J Biol Chem 2010; 285:25196 - 25202; PMID: 20554520; http://dx.doi.org/10.1074/jbc.M110.119446
- Basu A, et al. PLoS One 2011; 6:23919; PMID: 21886838; http://dx.doi.org/10.1371/journal.pone.0023919
- Sancak Y, et al. Mol Cell 2007; 25:903 - 915; PMID: 17386266; http://dx.doi.org/10.1016/j.molcel.2007.03.003
- Vander Haar E, et al. Nat Cell Biol 2007; 9:316 - 323; PMID: 17277771; http://dx.doi.org/10.1038/ncb1547
- Blagosklonny MV. Cancer Biol Ther 2008; 7:1520 - 1524; PMID: 18769112; http://dx.doi.org/10.4161/cbt.7.10.6663
- Pan F, et al. Nature 2007; 445:433 - 436; PMID: 17230191; http://dx.doi.org/10.1038/nature05476