Abstract
The deubiquitinating enzyme USP2a has shown oncogenic properties in many cancer types by impairing ubiquitination of FASN, MDM2, MDMX or Aurora A. Aberrant expression of USP2a has been linked to progression of human tumors, particularly prostate cancer. However, little is known about the role of USP2a or its mechanism of action in bladder cancer. Here, we provide evidence that USP2a is an oncoprotein in bladder cancer cells. Enforced expression of USP2a caused enhanced proliferation, invasion, migration and resistance to several chemotherapeutic reagents, while USP2a loss resulted in slower proliferation, greater chemosensitivity and reduced migratory/invasive capability compared with control cells. USP2a, but not a catalytically inactive mutant, enhanced proliferation in immortalized TRT-HU1 normal human bladder epithelial cells. USP2a bound to cyclin A1 and prevented cyclin A1 ubiquitination, leading to accumulation of cyclin A1 by a block in degradation. Enforced expression of wild type USP2a, but not an inactive USP2a mutant, resulted in cyclin A1 accumulation and increased cell proliferation. We conclude that USP2a impairs ubiquitination and stabilizes an important cell cycle regulator, cyclin A1, raising the possibility of USP2a targeting as a therapeutic strategy against bladder tumors in combination with chemotherapy.
Introduction
Bladder cancer is the second most common genitourinary malignancy in the United States and Europe, with 69,250 new cases and 14,990 deaths from bladder cancer in the US estimated in 2011.Citation1,Citation2 Bladder cancers present as transitional cell carcinoma (TCC), squamous cell carcinoma, adenocarcinoma and a few other rare subtypes. TCC is the most predominant histological type, which includes papillary, well- or moderately differentiated non-muscle invasive bladder cancer (NMIBC).Citation3 Cancer recurrence occurs in up to 70% of patients with NMIBC, and 20–30% of these patients show progression to muscle invasive bladder cancer and poor outcomes.Citation4,Citation5 Compared with other cancer types, bladder cancer is characteristically sensitive to cisplatin-based chemotherapy, which is a standard treatment.Citation6 Molecular mechanisms and clinical impact of combination therapy with other drugs based on targeted signaling strategies (e.g., Gefitinib, Trastuzumab, Cetuximab, Everolimus and Sorafenib) or angiogenesis inhibition (e.g., Sunitinib and Bevacizumab) are under investigation.Citation7–Citation11 Recent functional genetic screens identified several cisplatin response modulators, suggesting molecular mechanisms as well as several therapeutic candidates, including DNA damage response-related genes.Citation12 For example, the hsp90 inhibitor 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) selectively sensitized bladder cancers to cisplatin in a mouse model.Citation13
Ubiquitination and protein degradation mechanisms have been reported to participitate in regulation of oncoproteins and tumor suppressors, and this is currently an active area of investigation. Much effort has been made to explore the roles of ubiquitin protein E3 ligases in mediating ubiquitination of cancer-relevant proteins, including receptor tyrosine kinases (RTKs).Citation14,Citation15 Deubiquitinating enzymes (DUBs) de-conjugate ubiquitin from substrates and from ubiquitin chains, thus negatively regulating ubiquitination.Citation16–Citation20
Although studies of ubiquitination and deubiquitination of proteins remain challenging due to the limited methods that are currently applied,Citation21 recently, several reports have described chemotherapeutic strategies to avoid chemoresistance based on mechanisms of uniqitination and deubiquitination. Expression of the pro-survival protein, Mcl-1, which is regulated by ubiquitination/deubiquitination, regulates chemoresistance of cells deficient in the tumor suppressor Fbw7.Citation22 Neznanov et al. reported that proteotoxic stress-targeted therapy induces protein misfolding and enhances the antitumor effects of the proteasome inhibitor bortezomib.Citation23 De-ubiquitinating enzymes (DUBs) such as AMSHCitation24 and UBPYCitation25,Citation26 have been implicated in the increased level of oncoproteins, such as epidermal growth factor receptor (EGFR). One of the DUBs and a ubiquitin-specific protease 2 (USP2) isoform, USP2a, has been demonstrated to regulate expression levels of fatty acid synthase (FASN), murine double minute 2 (MDM2), MDMX and Aurora A,Citation27–Citation31 all well-known oncoproteins relevant to tumor progression in many cancer types, including bladder cancer.Citation32,Citation33 One USP2a target, FASN, plays a key role in lipid metabolism by synthesizing palmitate from acetyl-CoA and malonyl-CoA. In bladder cancer, FASN expression level was described as a predictor of recurrence and of tumor aggressiveness when combined with Ki-67 expression index.Citation34 A recently identified USP2a substrate,Citation30 Aurora A, is involved in regulation of centrosomal and microtubule activity and control of chromosome segregation;Citation35 thus, Aurora A overexpression leads to impaired chromosomal stability and aneuploidy. Aurora A, an oncogenic serine/threonine kinase, is overexpressed in bladder cancer tissues from patients as assessed by both RT-PCR and immunohistochemical assays.Citation36–Citation40 Taken together, these data suggest a role for USP2a as a mediator of bladder cancer progression.
Here, we show that USP2a associates with and stabilizes an important cell cycle regulator, cyclin A1, resulting in enhanced oncogenic characteristics expressed by bladder cancer cells. Our findings suggest that impaired ubiquitination and protein degradation in bladder tumor cells can be therapeutically targeted.
Results
Aberrant expression of USP2a enhances T24 cancer cell proliferation and invasion.
To determine whether the deubiquitinase USP2a plays a role in bladder cancer progression, we attempted to analyze the relationship between USP2a expression and oncogenesis. T24 human bladder cancer cells were transiently transfected with a wild-type USP2a construct (USP2aWT) or vector plasmid only (Vec). In experiments where transfected T24 bladder cancer cells were stimulated with growth medium after 16 h serum starvation, we observed that USP2aWT bladder cancer cells grew faster than control cells (). To independently assess the impact of USP2aWT on migration, a migration assay using gelatin-coated chambers was performed. Enforced USP2aWT enhanced migration about 2-fold, compared with control (). The total cell mass of USP2aWT cultures reached about 2-fold of control cells 1 d after stimulation (p < 0.05). Increased cell migration by enforced expression of USP2aWT was also observed in a wound-healing assay (). Colony formation capability as measured by in vitro clonogenic assay reports oncogenic properties. Approximately 10-fold more colonies (diameter >100 µm) were formed in USP2aWT bladder cancer cells, compared with control cells ().
These results suggest that human T24 cells expressing USP2aWT acquire a more aggressive phenotype, probably by activation of oncogenic signals. To determine the mechanism whereby this deubiquitinase would alter biological responses to growth stimulating factors, we assessed the activation status of several signal transduction pathways. Erk/MAPK is activated by the autocrine regulator of human bladder epithelial cells, heparin-binding EGF-like growth factor (HB-EGF).Citation41 HB-EGF has been reported to promote cell growth and induce phosphorylation of essential signaling pathways, including Erk/MAPK, Akt1 and p70S6K but not p38MAPK in bladder cells.Citation42 The extent of Erk/MAPK phosphorylation by HB-EGF treatment in USP2aWT cells was compared with control cells (Vec). Cells expressing USP2aWT responded more rapidly to HB-EGF stimulation 5 min after treatment and Erk/MAPK phosphorylation was sustained until 60 min, while control cells exhibited transient Erk/MAPK phosphorylation (). In order to assess the effect of USP2aWT on sensitivity to cisplatin, we used a clinically available chemotherapeutic drug that causes crosslinking of DNA and apoptosis induction; USP2aWT T24 cells were incubated in 10 µM cisplatin-containing medium for 24 h and compared with control cells. Treating the USP2aWT cells (and control cells) with 10 µM 12-O-tetradecanoylphorbol-13-acetate (TPA), 5 µM nocodazole (G2/M blocker) or 10 µM cycloheximide (CHX, protein synthesis inhibitor) for 24 h was followed by measurement of apoptotic markers to determine sensitivity to apoptosis inducers. TUNEL assay () and western blot analysis detecting the cleaved form of PARP (c-PARP) revealed that USP2aWT cells are more resistant to apoptosis induction by cisplatin, as well as other apoptosis inducers ().
USP2a loss attenuates proliferation and migration in T24 human bladder cancer cells.
Given the finding that USP2a may enhance oncogenic properties of bladder cancer, we performed similar experiments with USP2a-deficient T24 cells (shUSP2a) to determine proliferation, migration and apoptosis resistance and compared with control cells (shCtrl). USP2a-deficient bladder cancer cells were generated by stable transfection with shRNA directed against USP2a. Western blot analysis confirmed reduced expression of USP2a protein in the USP2A-silenced cell lines, T24-shUSP2a-c1 and T24-shUSP2a-c2, compared with T24-shCtrl cells (). Cell proliferation and migration were significantly suppressed by USP2a knockdown. These biological effects were consistent with the relative expression level of USP2a. The T24-shUSP2a-c2 cell line exhibited slower proliferation and migration, with a greater decrease of USP2a expression ( and C). Compared with control cells, USP2a-knockdown cells invaded more slowly through matrigel-coated filters (). We also observed that USP2a deficiency made T24 cells more sensitive to treatment with apoptotic reagents, including cisplatin ().
As in T24 bladder cancer cells, enforced expression of USP2aWT substantially enhanced colony formation in the immortalized bladder epithelial cell line TRT-HU1Citation43 (). However, inactive USP2a, USP2aMUT, did not show this pro-proliferative effect, suggesting that catalytic activity is important for USP2a function ().
Association of USP2a and cyclin A1 attenuated cyclin A1 expression.
The above experimental results suggest that enforced expression of USP2a in T24 cells, probably through suppression of ubiquitination of critical protein(s), leads to a more aggressive tumor cell phenotype. Because functional analysis of USP2a suggested that this deubiquitinase might control proliferation through regulation of cell cycle transition, we questioned whether enforced expression or loss of USP2a expression alters expression of cell cycle-related proteins. Blotting of T24-USP2aWT cell lysates with antibodies against cyclin A1 or cyclin D1 suggested that expression level of cell cycle regulator(s) positively correlates with USP2a level ( and B). USP2aWT-overexpressing T24 cells showed an increased expression of cell cycle regulators (), consistent with the observed enhanced proliferation (). USP2a knockdown by RNA interference (RNAi) using a pool of four siRNA oligos targeted to USP2a reversed the effect caused by USP2a overexpression (). Previously, an important cell cycle regulator, cyclin D1, was identified as a substrate of USP2, which consists of two splice variants, USP2a and USP2b.Citation44 In this study, we assessed whether USP2a regulates cyclin A1 by examining whether cyclin A1 directly binds to USP2a, thus resulting in protein accumulation. The cell cycle-regulatory protein cyclin A1 is required for G2/M progression during meitotic division of male germ cellsCitation45 and cell cycle transition in somatic cells.Citation46 As a proto-oncogene, levels of cyclin A1 expression correlate with patient outcome in various tumor types, including prostate cancer, breast cancer, acute myeloid leukemia and acute lymphoblastic leukemia.Citation47–Citation50 Further analysis using co-precipitation with anti-cyclin A1 antibody and immunoblotting with anti-USP2a antibody revealed that cyclin A1 physically interacts with USP2a (). The potential influence of USP2aWT on cyclin A1 stability was assessed by inhibiting protein synthesis with CHX. In the setting of enforced USP2aWT, cyclin A1 levels were sustained in the presence of CHX. Enforced expression of USP2aWT attenuated cyclin A1 degradation (). We conclude that USP2a regulates cyclin A1 at the posttranslational level.
Discussion
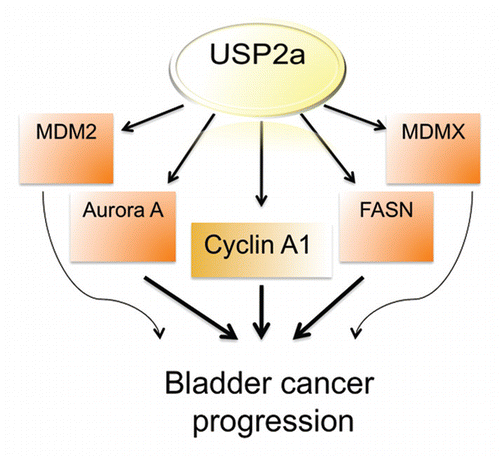
In this study, we show that increased cyclin A1 in human bladder cancer cells may be regulated by activity of the USP2a deubiquitinase. Our data suggest that USP2a is a pro-proliferative and pro-migratory protein relevant to features of clinically aggressive bladder cancer, consistent with observations in other cancer types. USP2a removes ubiquitin from protein chains, leading to alterations in the rate of ubiquitination-induced protein degradation. Since ubiquitination and resulting protein degradation is a dynamic and important mechanism in the tight control of protein expression in cells, a balance between ubiqutination and deubiquitination processes control protein degradation rates.Citation16,Citation18,Citation20,Citation51 In addition to the known function of USP2a as a deubiquiting enzyme, which is critical for regulated expression of FASN, MDM2, MDMX or Aurora A,Citation28–Citation30,Citation32 we conclude that USP2a is involved in cyclin A1 degradation, at least in part by a deubiquitination mechanism.
Cyclin A1, a context-dependent regulator of the G1/S- or G2/M-phase transitions,Citation45,Citation46 binds to cyclin dependent kinases 1/2 (CDK1/2) and activates downstream targets, including the retinoblastoma protein (Rb), E2F transcription factors and B-MYB (a cell cycle-associated transcription factor), leading to cell cycle transition.Citation52–Citation55 Cyclin A1-targeting experiments in the mouse showed that male cyclin A1-/- mice exhibit impaired spermatogenesis, and males are sterile, suggesting that cyclin A1 plays a critical role in fertility through regulation of the cell cycle.Citation56 Transgenic cyclin A1-overexpressing mice develop abnormal myelopoiesis and acute myeloid leukemia.Citation50 Cyclin A1 may be involved in pathogenesis of primary and metastatic cancer, because elevated cyclin A1 expression has been observed in various types of solid tumors, including testicular, ovarian, breast and prostate cancers.Citation47,Citation49,Citation50 Recent findings have suggested the underlying regulatory mechanism whereby cyclin A1 contributes to prostate cancer invasion. Cyclin A1 associates with androgen receptor (AR) and regulates the expression of matrix metalloproteases (MMPs) and vascular endothelial growth factor (VEGF), leading to a more invasive and metastatic phenotype.Citation47,Citation57 However, pathoclinical studies or functional analyses to understand the expression status or the precise role of cyclin A1 in primary and metastatic bladder cancers are limited. Our results suggest that further investigation into the mechanisms whereby cyclin A1 contributes to bladder cancer development and progression to advanced disease are warranted.
Our findings show that aberrant expression of USP2a significantly upregulated the proliferation and migration capability of T24 cells (), while USP2a knockdown suppressed those effects compared with controls (). These findings suggest the possibility of USP2a-suppressing reagents as an approach to cancer therapy. A recently reported synthetic compound with antioxidant activity, HO-3867, exhibited anticancer effects on many cancer types, including breast, colon, head and neck, liver, lung, ovarian and prostate cancers,Citation58 by significant downregulation of USP2a, causing apoptosis. Knockdown of USP2a enhanced the cisplatin sensitivity in testicular embryonal carcinoma cells.Citation29 This finding is suggestive of the possibility that clinical application of USP2a inhibitors to the cisplatin-resistant patient population may result in clinical benefit.
In summary, the present study provides evidence suggesting a role for the USP2a deubiquitinase in proliferation and migration of human bladder cancer cells. We showed that USP2a targets a cell cycle regulator, cyclin A1, thereby regulating cyclin A1 expression level, cell growth and apoptosis (). These findings provide a potential strategy in which the USP2a-cyclin A1 pathway might be pharmacologically manipulated as a means of bladder cancer therapy.
Experimental Procedures
Reagents.
Dialyzed fetal bovine serum (FBS), Dulbecco's Modified Eagle Medium (DMEM) and Lipofectamine 2000 were from Invitrogen. Micro BCA protein assay kit was obtained from Pierce. Coomassie Blue R-250 staining solution and destaining solution were from Bio-Rad. Small interfering RNAs (siRNAs) against USP2a and OFF-TARGET controls were from Dharmacon. Protease inhibitor cocktail tablets were from Roche Diagnostics. The following antibodies were used: antibodies to USP2a (Millennium Pharmaceuticals, Inc., and/or ABGENT), cyclin D1 (Biosource), cyclin A1 (Abcam Inc.), ubiquitin (Calbiochem), FASN (Santa Cruz Biotechnology), EGFR (Invitrogen), β-actin (Sigma-Aldrich). All other reagents were highest quality and were obtained from Sigma-Aldrich or Promega.
Cell culture and cell transfection.
T24 cells (ATCC) were cultured in DMEM supplemented with 10% FBS, 100 units/mL penicillin and 100 µg/mL streptomycin at 37°C in a humidified incubator with 5% CO2. Immortalized human normal bladder epithelial TRT-HU1 cells were recently described in reference Citation43. For transient transfection, cells were grown to 80% confluence, and transfection was performed with Lipofectamine 2000 using 1 µg plasmid DNA. Wild-type USP2a (USP2aWT) or catalytically inactive USP2a (USP2aMUT) were cloned into pEGFP and used for transfection. Catalytic Cys was mutated to Ala in USP2aMUT (C276A). For silencing, cells were transfected with 80 nmol/L of anti-USP2a siRNA [r(UGC UUG UGC CCG GUU CGA C)d(TT)]. OFF-TARGET siRNAs were used as control. To generate stable knockout cell lines, T24 cells were transfected with shVec or shUSP2a, followed by selection with G418 for 6 weeks. Two clones showing maximum knockdown, shUSP2a-c1 and shUSP2a-c2, were chosen and used for the experiments described.
Western blot analysis.
Whole-cell lysates were prepared with lysis buffer (1% Nonidet P-40, 50 mM Tris pH 7.4, 10 mM NaCl, 1 mM NaF, 5 mM MgCl2, 0.1 mM EDTA, 1 mM PMSF, protease inhibitor cocktail) and centrifuged at 12,000x g for 15 min. Protein concentrations were determined by Micro BCA assay according to the manufacturer's protocol. Proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes for immunoblotting analysis. To standardize protein levels, the blots were reprobed with antibodies against β-actin.
Cell proliferation assay.
T24 human bladder cancer cells were plated onto 6-well culture plates at a density of 1 × 103 cells per well in standard DMEM growth medium with 10% FBS. When cells grew to ∼80% confluence, they were transfected with expression constructs or siRNAs. Three days after transfection, cell proliferation rate was determined by crystal violet staining.Citation59 Absorbance was measured at 570 nm.
Migration and wound-healing assay.
Matrigel-coated inserts (Millipore Corp.) were rehydrated before seeding T24 cells at 3 × 105 cells/ml on the upper surface of inserts. Sixteen h after seeding, non-migratory cells in interior surface of inserts were removed. Invaded cells on the bottom surface of the inserts were stained with the cell staining solution and counted. Eluates with 10% acetic acid based extraction solution were transferred to 96-well microtiter plates and absorbance read at 560 nm using a FLUOstar plate reader. Wound-healing assays were performed in 6-well plates after cell density reached confluence. After scratching cells with a sharp tip, the plates were returned to the tissue culture incubator for 16 h before fixation and imaging.
Colony formation assay.
Transfected cells were seeded in 150 mm plate at 1 × 102/well. Cells were fixed and stained with crystal violet reagents for counting at 7 d.
Apoptosis analysis.
Apoptosis was determined by two independent methods, the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay and western blotting with anti-c-PARP antibody. For TUNEL assay (Roche Applied Science), reaction mixture or negative control solution was applied to each slide after cell fixation using 4% paraformaldehyde. Detection of apoptotic cells and counterstaining with propidium iodide (PI) were performed according to the instructions provided by the manufacturer. TUNEL positivity was counted and analyzed under an Axioplan 2 microscope (Carl Zeiss MicroImaging, Inc).
Co-immunoprecipitation.
Whole-cell lysates from T24 cells were prepared with lysis buffer containing 50 mM HEPES pH 7.3, 10% glycerol, 1% Triton X-100, 0.5 mM Na3VO4, protease inhibitor tablet (Roche), 10 mM NEM. Lysate was then incubated with protein A/G-Sepharose beads pre-loaded with antibodies against cyclin A1 or USP2a for 4 h in a cold room. Immunoprecipitates were washed twice with lysis buffer and twice with lysis buffer containing 500 mM NaCl. The precipitates were resuspended in 2x SDS loading buffer for western blot analysis.
Cyclin A1 degradation.
T24 cells transiently transfected with an USP2aWT (or Vec) were incubated with or without 10 µg/ml cycloheximide (CHX). After treatment for 15, 30 or 45 min, whole-cell lysates were prepared and applied for western blot with anti-cyclin A1 antibody.
Statistical analysis.
Statistical analysis was performed using data from at least 3 independent experiments and at least 3 samples per experiment. Data are shown as mean ± standard deviation (SD) and statistical significance of differences between means was assessed by two-tailed Student t-test, and p < 0.05 was considered statistically significant.
Author Contributions
J. K. and M.R.F. conceived and designed experiments and wrote the paper. W.K., Z.L. and M.F.L. contributed reagents, materials, expertise related to bladder cancer and USP2a and participated data analysis. J.K., W.K., M.F.L. and M.R.F. interpreted data as well as helped to prepare and revise the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| 17-AAG | = | 17-(allylamino)-17-demethoxygeldanamycin |
| CHX | = | cycloheximide |
| c-PARP | = | cleaved form of poly (ADP-ribose) polymerase |
| DMEM | = | dulbecco's modified eagle medium |
| DUBs | = | deubiquitinating enzymes |
| EGFR | = | epidermal growth factor receptor |
| FASN | = | fatty acid synthase |
| FBS | = | fetal bovine serum |
| HB-EGF | = | heparin-binding EGF-like growth factor |
| MDM2 | = | murine double minute 2 |
| MMPs | = | matrix metallopeptidase |
| NMIBC | = | non-muscle invasive bladder cancer |
| RNAi | = | RNA interference |
| RTKs | = | receptor tyrosine kinases |
| siRNAs | = | small interfering RNAs |
| TCC | = | transitional cell carcinoma |
| TPA | = | 12-O-tetradecanoylphorbol-13-acetate |
| TUNEL | = | deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling |
| USP2a | = | ubiquitin-specific protease 2a |
| VEGF | = | vascular endothelial growth factor |
Figures and Tables
Figure 1 Enforced USP2aWT expression enhanced tumorigenesis in T24 human bladder cancer cells. (A) Proliferation rate in T24 bladder cancer cells transiently engineered to express USP2aWT, T24-USP2aWT and Vec (vector only), T24-Vec. Cells were serum starved for 16 h before changing into growth medium. At the indicated time points, cells were fixed and stained with crystal violet solution. (B) Cell migration capability of T24-USP2aWT and T24 Vec cells were analyzed using gelatin-coated invasion chamber kit (company) as described in the Materials and Methods section. (C) Migration was compared in T24-USP2aWT and T24 Vec cells by a wound-healing assay. (D) T24-USP2aWT and T24-Vec cells were seeded at low density and incubated for colony formation for 7 d. After fixing and staining cells, number of colonies was counted. Representative images were shown. Asterisks in this figure indicate p < 0.05.
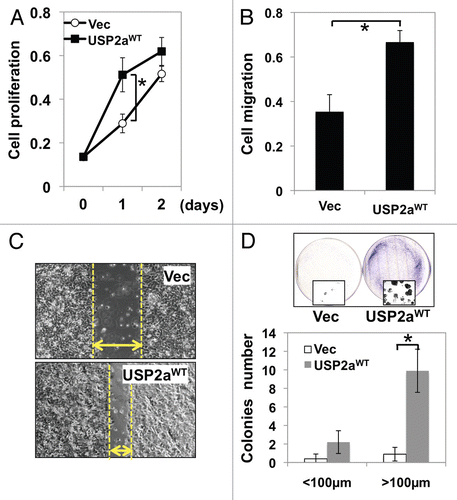
Figure 2 USP2aWT altered activation of signal transduction. (A) Erk/MAPK phosphorylation in USP2aWT expressing T24 cells treated with recombinant HB-EGF (100 ng/ml). Protein from whole-cell lysates at the indicated times were blotted with the Abs against p-Erk/MAPK, Erk/MAPK and β-actin. (B) TUNEL assay showed less apoptotic cells in USP2a-expressing cells compared with control. (C) Levels of cleaved PARP (c-PARP) were detected in whole lysates from T24-USP2aWT and T24-Vec cells incubated in serum free medium (SF), TPA, nocodazole, cisplatin or CHX for 24 h.
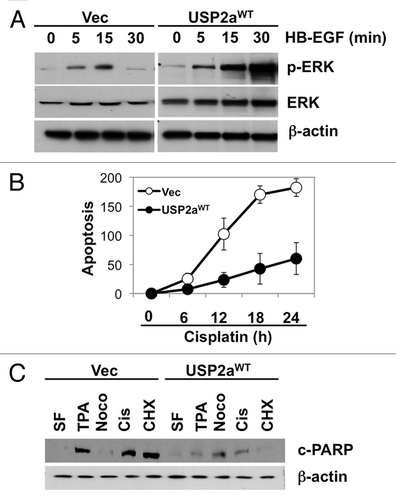
Figure 3 USP2a knockdown by shRNA suppressed oncogenesis. (A) Verification of USP2a gene silencing by stable transfection of T24 cells using two kinds of shUSP2a. USP2a expression level in two engineered cells, shUSP2a-c1 and shUSP2a-c2, was analyzed by western blotting. Control shRNA (shCtrl) was used as negative control. (B) Proliferation of two USP2a-knockdown cell lines was compared with control cells. (C) Wound-healing assay to assess migratory capability of USP2a loss shUSP2a-c2 and control cells. (D) Quantitative analysis using invasion chamber kit of cell migration in USP2a loss cells. (E) USP2a-knockdown cells are more susceptible to apoptotic inducers including a chemotherapeutic drug, cisplatin. Stably engineered cell lines were treated with the indicated reagents (see the Materials and Methods), and protein extracts were applied for western blotting with anti-c-PARP antibody. Asterisks indicate p < 0.05.
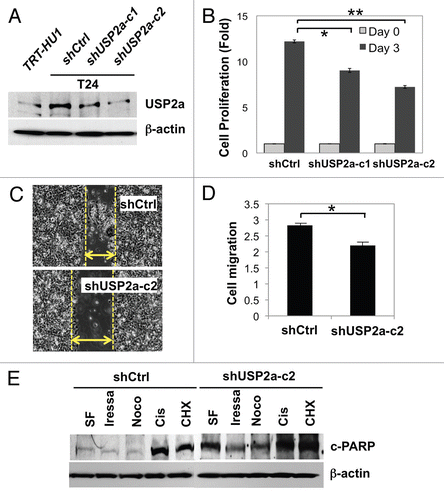
Figure 4 USP2a enhanced the proliferation rate of immortalized human bladder epithelial cells (TRT-HU1) via catalytic activity. (A) A greater colonization by USP2a overexpression in TRT-HU1 cells. Cells were seeded at low density (102/dish) and incubated for 14 d. Colonies were counted after crystal violet staining. (B) Effect of a mutant form of USP2a on proliferation in TRT-HU1 cells. Cell proliferation was evaluated using crystal violet.
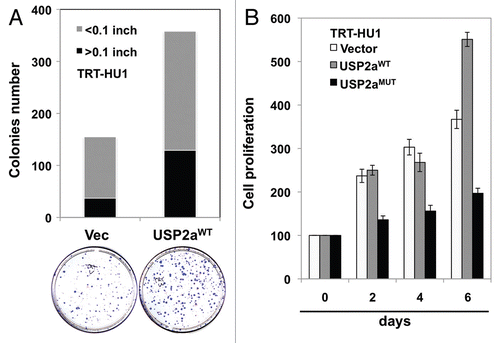
Figure 5 USP2a stabilized cyclin A1. (A) USP2aWT expressing T24 cells expressed higher level of cyclin D1 and cyclin A1. (B) USP2a-targeted oligos, siUSP2a, reversed an increase of cyclin D1 and cyclin A1. (C) Physical interaction of USP2a and cyclin A1. Whole-cell lysates (200 µg) from T24 cells were applied to immunoprecipitation (IP) with anti-cyclin A1 antibody, and IP product was blotted with anti-USP2a antibody (∼70 kDa). The reciprocal co-immunoprecipitation assay was not applied due to no difference between cyclin A1 and IgG (50–55 kDa), although the IP itself was working and we could detect EGFR and FASN co-immunoprecipitated with USP2a. (D) Cyclin A1 degradation was measured in absence or presence of 10 µg/ml CHX in the T24-USP2aWT and T24-Vec cells.
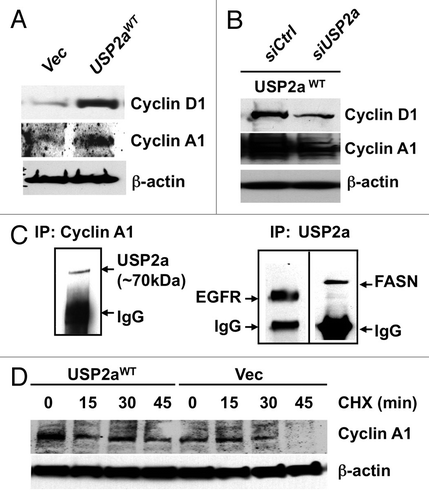
Figure 6 Diagram showing potential USP2a signal network during bladder cancer progression.
Acknowledgements
We thank Dr. Leland W. Chung (Cedars-Sinai Medical Center) for critical review of the manuscript and helpful advice. This work was supported by the following funding sources: NIH grants R01 DK087806, R01 CA143777 and P50 DK65298 (to M.R.F.); the Fishbein Family IC Research Foundation/Interstitial Cystitis Association (ICA), Pilot Research Program/ICA, New York Academy of Medicine and Children's Hospital Boston Faculty Development (to J.K.). J.K. is an American Urological Association Foundation Research Scholar and a Harvard Medical School Eleanor and Miles Shore Scholar.
Related Research Data
References
- Kirkali Z, Chan T, Manoharan M, Algaba F, Busch C, Cheng L, et al. Bladder cancer: epidemiology, staging and grading, and diagnosis. Urology 2005; 66:4 - 34; PMID: 16399414; http://dx.doi.org/10.1016/j.urology.2005.07.062
- Babjuk M, Oosterlinck W, Sylvester R, Kaasinen E, Böhle A, Palou-Redorta J, European Association of Urology (EAU). EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder. Eur Urol 2008; 54:303 - 314; PMID: 18468779; http://dx.doi.org/10.1016/j.eururo.2008.04.051
- Kiemeney LA, Witjes JA, Verbeek AL, Heijbroek RP, Debruyne FM, Dutch South-East Cooperative Urological Group. The clinical epidemiology of superficial bladder cancer. Br J Cancer 1993; 67:806 - 812; PMID: 8471440; http://dx.doi.org/10.1038/bjc.1993.147
- Borden LS Jr, Clark PE, Hall MC. Bladder cancer. Curr Opin Oncol 2003; 15:227 - 233; PMID: 12778017; http://dx.doi.org/10.1097/00001622-200305000-00009
- Hall MC, Chang SS, Dalbagni G, Pruthi RS, Seigne JD, Skinner EC, et al. Guideline for the management of nonmuscle invasive bladder cancer (stages Ta, T1 and Tis): 2007 update. J Urol 2007; 178:2314 - 2330; PMID: 17993339; http://dx.doi.org/10.1016/j.juro.2007.09.003
- Cognetti F, Ruggeri EM, Felici A, Gallucci M, Muto G, Pollera CF, et al. Adjuvant chemotherapy with cisplatin and gemcitabine versus chemotherapy at relapse in patients with muscle-invasive bladder cancer submitted to radical cystectomy: an Italian, multicenter, randomized phase III trial. Ann Oncol 2011; on behalf of the Study Group Epub ahead of print PMID: 21859900; http://dx.doi.org/10.1093/annonc/mdr354
- Beekman KW, Bradley D, Hussain M. New molecular targets and novel agents in the treatment of advanced urothelial cancer. Semin Oncol 2007; 34:154 - 164; PMID: 17382799; http://dx.doi.org/10.1053/j.seminoncol.2006.12.007
- Black PC, Agarwal PK, Dinney CP. Targeted therapies in bladder cancer—an update. Urol Oncol 2007; 25:433 - 438; PMID: 17826665; http://dx.doi.org/10.1016/j.urolonc.2007.05.011
- Black PC, Dinney CP. Growth factors and receptors as prognostic markers in urothelial carcinoma. Curr Urol Rep 2008; 9:55 - 61; PMID: 18366975; http://dx.doi.org/10.1007/s11934-008-0011-6
- Liou LS. Urothelial cancer biomarkers for detection and surveillance. Urology 2006; 67:25 - 33; PMID: 16530072; http://dx.doi.org/10.1016/j.urology.2006.01.034
- Said N, Theodorescu D. Pathways of metastasis suppression in bladder cancer. Cancer Metastasis Rev 2009; 28:327 - 333; PMID: 20013033; http://dx.doi.org/10.1007/s10555-009-9197-4
- Nijwening JH, Kuiken HJ, Beijersbergen RL. Screening for modulators of cisplatin sensitivity: unbiased screens reveal common themes. Cell Cycle 2011; 10:380 - 386; PMID: 21239890; http://dx.doi.org/10.4161/cc.10.3.14642
- Yoshida S, Koga F, Tatokoro M, Kawakami S, Fujii Y, Kumagai J, et al. Low-dose Hsp90 inhibitors tumor-selectively sensitize bladder cancer cells to chemoradiotherapy. Cell Cycle 2011; 10:4291 - 4299; PMID: 22134243; http://dx.doi.org/10.4161/cc.10.24.18616
- d'Azzo A, Bongiovanni A, Nastasi T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 2005; 6:429 - 441; PMID: 15882441; http://dx.doi.org/10.1111/j.1600-0854.2005.00294.x
- Komada M. Controlling receptor downregulation by ubiquitination and deubiquitination. Curr Drug Discov Technol 2008; 5:78 - 84; PMID: 18537571; http://dx.doi.org/10.2174/157016308783769469
- Hussain S, Zhang Y, Galardy PJ. DUBs and cancer: the role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 2009; 8:1688 - 1697; PMID: 19448430; http://dx.doi.org/10.4161/cc.8.11.8739
- Love KR, Catic A, Schlieker C, Ploegh HL. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat Chem Biol 2007; 3:697 - 705; PMID: 17948018; http://dx.doi.org/10.1038/nchembio.2007.43
- Niedzwiedz W, Patel KJ. “Dub” bing a tumor suppressor pathway. Cancer Cell 2005; 7:114 - 115; PMID: 15710323; http://dx.doi.org/10.1016/j.ccr.2005.01.018
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005; 123:773 - 786; PMID: 16325574; http://dx.doi.org/10.1016/j.cell.2005.11.007
- Singhal S, Taylor MC, Baker RT. Deubiquitylating enzymes and disease. BMC Biochem 2008; 9:3; PMID: 19007433; http://dx.doi.org/10.1186/1471-2091-9-S1-S3
- Stringer DK, Piper RC. Terminating protein ubiquitination: Hasta la vista, ubiquitin. Cell Cycle 2011; 10:3067 - 3071; PMID: 21926471; http://dx.doi.org/10.4161/cc.10.18.17191
- Inuzuka H, Fukushima H, Shaik S, Liu P, Lau AW, Wei W. Mcl-1 ubiquitination and destruction. Oncotarget 2011; 2:239 - 244; PMID: 21608150
- Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget 2011; 2:209 - 221; PMID: 21444945
- Ma YM, Boucrot E, Villén J, Affar B, Gygi SP, Göttlinger HG, et al. Targeting of AMSH to endosomes is required for epidermal growth factor receptor degradation. J Biol Chem 2007; 282:9805 - 9812; PMID: 17261583; http://dx.doi.org/10.1074/jbc.M611635200
- Naviglio S, Mattecucci C, Matoskova B, Nagase T, Nomura N, Di Fiore PP, et al. UBPY: a growth-regulated human ubiquitin isopeptidase. EMBO J 1998; 17:3241 - 3250; PMID: 9628861; http://dx.doi.org/10.1093/emboj/17.12.3241
- Mizuno E, Iura T, Mukai A, Yoshimori T, Kitamura N, Komada M. Regulation of epidermal growth factor receptor downregulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell 2005; 16:5163 - 5174; PMID: 16120644; http://dx.doi.org/10.1091/mbc.E05-06-0560
- Priolo C, Tang D, Brahamandan M, Benassi B, Sicinska E, Ogino S, et al. The isopeptidase USP2a protects human prostate cancer from apoptosis. Cancer Res 2006; 66:8625 - 8632; PMID: 16951176; http://dx.doi.org/10.1158/0008-5472.CAN-06-1374
- Stevenson LF, Sparks A, Allende-Vega N, Xirodimas DP, Lane DP, Saville MK. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J 2007; 26:976 - 986; PMID: 17290220; http://dx.doi.org/10.1038/sj.emboj.7601567
- Allende-Vega N, Sparks A, Lane DP, Saville MK. MdmX is a substrate for the deubiquitinating enzyme USP2a. Oncogene 2010; 29:432 - 441; PMID: 19838211; http://dx.doi.org/10.1038/onc.2009.330
- Shi Y, Solomon LR, Pereda-Lopez A, Giranda VL, Luo Y, Johnson EF, et al. The deubiquitinase USP2A regulates the stability of Aurora A. J Biol Chem 2011; 286:38960 - 38968; PMID: 21890637; http://dx.doi.org/10.1074/jbc.M111.231498
- Inuzuka H, Fukushima H, Shaik S, Wei W. Novel insights into the molecular mechanisms governing Mdm2 ubiquitination and destruction. Oncotarget 2010; 1:685 - 690; PMID: 21317463
- Graner E, Tang D, Rossi S, Baron A, Migita T, Weinstein LJ, et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 2004; 5:253 - 261; PMID: 15050917; http://dx.doi.org/10.1016/S1535-6108(04)00055-8
- da Silva SD, Cunha IW, Nishimoto IN, Soares FA, Carraro DM, Kowalski LP, et al. Clinicopathological significance of ubiquitin-specific protease 2a (USP2a), fatty acid synthase (FASN), and ErbB2 expression in oral squamous cell carcinomas. Oral Oncol 2009; 45:134 - 139; PMID: 19362044; http://dx.doi.org/10.1016/j.oraloncology.2009.02.004
- Sugino T, Baba K, Hoshi N, Aikawa K, Yamaguchi O, Suzuki T. Overexpression of fatty acid synthase in human urinary bladder cancer and combined expression of the synthase and Ki-67 as a predictor of prognosis of cancer patients. Med Mol Morphol 2011; 44:146 - 150; PMID: 21922386; http://dx.doi.org/10.1007/s00795-010-0517-0
- Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link?. Trends Cell Biol 2005; 15:241 - 250; PMID: 15866028; http://dx.doi.org/10.1016/j.tcb.2005.03.004
- Liu HS, Ke CS, Cheng HC, Huang CY, Su CL. Curcumin-induced mitotic spindle defect and cell cycle arrest in human bladder cancer cells occurs partly through inhibition of aurora A. Mol Pharmacol 2011; 80:638 - 646; PMID: 21757545; http://dx.doi.org/10.1124/mol.111.072512
- Fraizer GC, Diaz MF, Lee IL, Grossman HB, Sen S. Aurora-A/STK15/BTAK enhances chromosomal instability in bladder cancer cells. Int J Oncol 2004; 25:1631 - 1639; PMID: 15547700
- Park HS, Park WS, Bondaruk J, Tanaka N, Katayama H, Lee S, et al. Quantitation of Aurora kinase A gene copy number in urine sediments and bladder cancer detection. J Natl Cancer Inst 2008; 100:1401 - 1411; PMID: 18812553; http://dx.doi.org/10.1093/jnci/djn304
- Tseng YS, Tzeng CC, Huang CY, Chen PH, Chiu AW, Hsu PY, et al. Aurora-A overexpression associates with Ha-ras codon-12 mutation and blackfoot disease endemic area in bladder cancer. Cancer Lett 2006; 241:93 - 101; PMID: 16338065; http://dx.doi.org/10.1016/j.canlet.2005.10.014
- Lei Y, Yan S, Ming-De L, Na L, Rui-Fa H. Prognostic significance of Aurora-A expression in human bladder cancer. Acta Histochem 2011; 113:514 - 518; PMID: 20598352; http://dx.doi.org/10.1016/j.acthis.2010.05.004
- Freeman MR, Yoo JJ, Raab G, Soker S, Adam RM, Schneck FX, et al. Heparin-binding EGF-like growth factor is an autocrine growth factor for human urothelial cells and is synthesized by epithelial and smooth muscle cells in the human bladder. J Clin Invest 1997; 99:1028 - 1036; PMID: 9062361; http://dx.doi.org/10.1172/JCI119230
- Kim J, Keay SK, Freeman MR. Heparin-binding epidermal growth factor-like growth factor functionally antagonizes interstitial cystitis antiproliferative factor via mitogen-activated protein kinase pathway activation. BJU Int 2009; 103:541 - 546; PMID: 18990151; http://dx.doi.org/10.1111/j.1464-410X.2008.08097.x
- Kim J, Ji M, DiDonato JA, Rackley RR, Kuang M, Sadhukhan PC, et al. An hTERT-immortalized human urothelial cell line that responds to anti-proliferative factor. In Vitro Cell Dev Biol Anim 2011; 47:2 - 9; PMID: 21136194; http://dx.doi.org/10.1007/s11626-010-9350-y
- Shan J, Zhao W, Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol Cell 2009; 36:469 - 476; PMID: 19917254; http://dx.doi.org/10.1016/j.molcel.2009.10.018
- Liu D, Liao C, Wolgemuth DJ. A role for cyclin A1 in the activation of MPF and G2-M transition during meiosis of male germ cells in mice. Dev Biol 2000; 224:388 - 400; PMID: 10926775; http://dx.doi.org/10.1006/dbio.2000.9776
- Ji P, Agrawal S, Diederichs S, Bäumer N, Becker A, Cauvet T, et al. Cyclin A1, the alternative A-type cyclin, contributes to G1/S cell cycle progression in somatic cells. Oncogene 2005; 24:2739 - 2744; PMID: 15829981; http://dx.doi.org/10.1038/sj.onc.1208356
- Wegiel B, Bjartell A, Tuomela J, Dizeyi N, Tinzl M, Helczynski L, et al. Multiple cellular mechanisms related to cyclin A1 in prostate cancer invasion and metastasis. Natl Cancer Inst 2008; 100:1022 - 1036; PMID: 18612129; http://dx.doi.org/10.1093/jnci/djn214
- Mirza A, Basso A, Black S, Malkowski M, Kwee L, Pachter JA, et al. RNA interference targeting of A1 receptor-overexpressing breast carcinoma cells leads to diminished rates of cell proliferation and induction of apoptosis. Cancer Biol Ther 2005; 4:1355 - 1360; PMID: 16294023; http://dx.doi.org/10.4161/cbt.4.12.2196
- Holm C, Ora I, Brunhoff C, Anagnostaki L, Landberg G, Persson JL. Cyclin A1 expression and associations with disease characteristics in childhood acute lymphoblastic leukemia. Leuk Res 2006; 30:254 - 261; PMID: 16182364; http://dx.doi.org/10.1016/j.leukres.2005.07.010
- Liao C, Wang XY, Wei HQ, Li SQ, Merghoub T, Pandolfi PP, et al. Altered myelopoiesis and the development of acute myeloid leukemia in transgenic mice overexpressing cyclin A1. Proc Natl Acad Sci USA 2001; 98:6853 - 6858; PMID: 11381140; http://dx.doi.org/10.1073/pnas.121540098
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 2009; 78:363 - 397; PMID: 19489724; http://dx.doi.org/10.1146/annurev.biochem.78.082307.091526
- Yang R, Morosetti R, Koeffler HP. Characterization of a second human cyclin A that is highly expressed in testis and in several leukemic cell lines. Cancer Res 1997; 57:913 - 920; PMID: 9041194
- Sweeney C, Murphy M, Kubelka M, Ravnik SE, Hawkins CF, Wolgemuth DJ, et al. A distinct cyclin A is expressed in germ cells in the mouse. Development 1996; 122:53 - 64; PMID: 8565853
- Yang R, Müller C, Huynh V, Fung YK, Yee AS, Koeffler HP. Functions of cyclin A1 in the cell cycle and its interactions with transcription factor E2F-1 and the Rb family of proteins. Mol Cell Biol 1999; 19:2400 - 2407; PMID: 10022926
- Müller-Tidow C, Wang W, Idos GE, Diederichs S, Yang R, Readhead C, et al. Cyclin A1 directly interacts with B-myb and cyclin A1/cdk2 phosphorylate B-myb at functionally important serine and threonine residues: tissue-specific regulation of B-myb function. Blood 2001; 97:2091 - 2097; PMID: 11264176; http://dx.doi.org/10.1182/blood.V97.7.2091
- Wolgemuth DJ. Function of the A-type cyclins during gametogenesis and early embryogenesis. Results Probl Cell Differ 2011; 53:391 - 413; PMID: 21630154; http://dx.doi.org/10.1007/978-3-642-19065-0_17
- Wegiel B, Bjartell A, Ekberg J, Gadaleanu V, Brunhoff C, Persson JL. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene 2005; 24:6385 - 6393; PMID: 16007189
- Selvendiran K, Ahmed S, Dayton A, Ravi Y, Kuppusamy ML, Bratasz A, et al. HO-3867, a synthetic compound, inhibits the migration and invasion of ovarian carcinoma cells through downregulation of fatty acid synthase and focal adhesion kinase. Mol Cancer Res 2010; 8:1188 - 1197; PMID: 20713491; http://dx.doi.org/10.1158/1541-7786.MCR-10-0201
- Yang W, Chung YG, Kim Y, Kim TK, Keay SK, Zhang CO, et al. Quantitative proteomics identifies a beta-catenin network as an element of the signaling response to Frizzled-8 protein-related antiproliferative factor. Mol Cell Proteomics 2011; 10:110; PMID: 21422242; http://dx.doi.org/10.1074/mcp.M110.007492