Abstract
Upon genotoxic stress and during normal S phase, ATM phosphorylates the checkpoint clamp protein Rad9 in a manner that depends on Ser272. Ser272 is the only known ATM-dependent phosphorylation site in human Rad9. However, Ser272 phosphorylation is not required for survival or checkpoint activation after DNA damage. The physiological function of Ser272 remains elusive. Here, we show that ATM-dependent Rad9Ser272 phosphorylation requires the MRN complex and controls repair pathways. Furthermore, the mutant cells accumulate large numbers of chromosome breaks and induce gross chromosomal rearrangements. Our findings establish a new and unexpected role for ATM: it phosphorylates the checkpoint clamp in order to control repair pathways, thereby maintaining genomic integrity during unperturbed cell cycle and upon DNA damage.
Keywords: :
Introduction
Cells protect themselves from genotoxic stresses by activating the cell cycle checkpoints and DNA repair pathways. Coordination of these pathways needs to be tightly regulated in order to prevent genomic instability.Citation1-Citation3 Mutations in checkpoint and repair genes can cause various diseases including human cancers.Citation4-Citation6 ATM (ataxia-telangiectasia-mutated) and ATR (ataxia telangiectasia and Rad3-related) kinases belong to the phosphatidylinositol 3-kinase-related kinase (PI-3 kinase) family, and are critical components of checkpoint activation. ATM and ATR kinase cascades lead to activation of downstream targets such as Brca1, Chk1, Chk2, p53 and Nbs1 in order to regulate cell cycle progression, apoptosis and DNA repair. ATM pathways are initiated primarily by double-strand breaks (DSBs),Citation7 while ATR responds to a broad spectrum of DNA damage and replication disruption, especially during S phase.Citation8
Choreography of the DNA damage response pathways has been shown in response to DSBs in budding yeast. The MRE11 and the ATM-related Tel1 kinase are the first proteins detected at DSBs. Next, the replication protein A (RPA) single-strand DNA (ssDNA) binding protein relocalizes to the break and recruits checkpoint proteins, such as RAD24 (Rad17 homolog), DDC1 (Rad9 homolog) and DDC2 (ATRIP homolog). Later and only in S and G2 phase, the homologs recombination proteins assemble at the site. Although the checkpoint clamp loader and clamp are recruited to the site before the homologs recombination proteins, they are not required for the recruitment of the homologs recombination proteins.Citation9 It is also shown in mammalian cells that ATM and MRE11 nuclease generate RPA coated ssDNA and ATR is recruited to DSB sites subsequently. MRE11 is required for ATM activation.Citation10-Citation13 ATM-dependent ATR activation in response to DSBs is restricted to the S and G2 cell cycle phases.Citation14 Other components of the ATR pathway, such as the checkpoint clamp complex (Rad9-Rad1-Hus1), are imprecated in regulating repair pathways in both mammals and yeasts.Citation1,Citation15 Function of the checkpoint clamp complex in response to DSBs is still largely unknown.
Numerous substrates of ATM and ATR kinases have been identified, including Rad9.Citation16-Citation22 Several phosphorylation sites on human Rad9 have been mapped.Citation23,Citation24 Among these sites, only Ser272 becomes phosphorylated in response to DNA damage. Moreover, it does so in an ATM-dependent manner. This is unexpected given that the checkpoint clamp complex (Rad9-Rad1-Hus1) is required for ATR-dependent, rather than ATM-dependent signaling.Citation25,Citation26 What is the function of this ATM-dependent Rad9 phosphorylation?
Results
ATM phosphorylates Rad9Ser272 upon damage, but phosphorylation is not required for survival or checkpoint activation upon DNA damage
To elucidate the function of the Rad9Ser272 phosphorylation, we created human Rad9-S272C and Rad9-S272A mutant cell lines. The mutant cell lines stably express exogenous Rad9-S272C and/or Rad9-S272A mutant proteins, and endogenous Rad9 expression is stably knocked down by its shRNA on the 3′ UTR region (hereafter Rad9-S272C and Rad9-S272A cell lines). We also created cell lines (hereafter Rad9 cell lines) in which exogenous wild type Rad9 proteins are stably expressed and endogenous Rad9 expression is stably knocked down (Fig. S1A). All experiments were performed under this condition. Rad9 knockdown cells showed defects in Chk1 activation after IR (ionizing radiation), UV (UV radiation), CPT (camptothecin, a topoisomerase I inhibitor) and HU (hydroxyurea). Chk2 activation in Rad9 knockdown cells was defective after UV, CPT and HU but not IR (Fig. S4). These results are consistent with previous observations that Rad9 is not required for ATM-dependent Chk2 activation after IR.Citation25 Chk1 and Chk2 became phosphorylated in response to damage in the Rad9-S272C and Rad9-S272A mutant cells, indicating that checkpoint responses of the mutant cells to DNA damage including IR, UV, CPT and HU were normal (Fig. S4, data not shown). No significant sensitivity to these DNA damaging agents was detected in both proliferation and clonogenic survival assays, although Ser272 became phosphorylated (Fig. S4 and S5). In fact, the Rad9-S272C cells showed slightly better survival after IR, UV and CPT treatments compared with the wild type control (p < 0.001, student’s t-test) (Fig. S5). Furthermore, damage-induced phosphorylation was abolished by an ATM-specific inhibitor KU55933 as well as two independent ATM siRNAs, indicating that damage-induced Ser272 phosphorylation depends on ATM ( and Fig. S1B).Citation27 Importantly, damage-induced Ser272 phosphorylation was abolished in MRE11, Rad50 and Nbs1 knockdown cells, indicating that damage-induced phosphorylation requires ATM activation ().
Figure 1. Rad9Ser272 is phosphorylated in an ATM-dependent manner. (A) Rad9Ser272 is phosphorylated in response to DNA damage. Rad9 becomes phosphorylated at Ser272 in response to IR, UV, CPT (6 h) and HU (1 h) and phosphorylation is inhibited by an ATM specific inhibitor KU55933 (10 µM) (left panel) and ATM specific siRNA (right panel). (B) Rad9Ser272 phosphorylation is not affected in ATR-Seckel cells after damage.
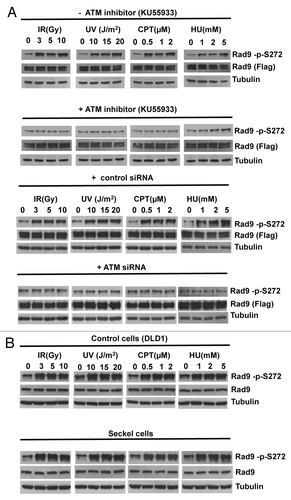
Figure 2. Rad9Ser272 is phosphorylated in a MRN complex-dependent manner. Damage-induced Ser272 phosphorylation is inhibited by siRNAs of MRE11, Rad50 and Nbs1.
UV and HU treatments are known to activate mainly the ATR-dependent pathway.Citation28,Citation29 However, UV and HU-induced phosphorylation was abolished or significantly reduced by the ATM-specific inhibitor and ATM siRNAs (). Damage induced Rad9Ser272 phosphorylation was not affected by ATR-specific siRNAs and in ATR-Seckel cells (; Fig. S2A). ATR seckel cells express very low level of ATR protein, and are shown to be defective in phosphorylating its substrates.Citation30,Citation31 Furthermore, knockdown of another PI3-kinase DNA-PKc by its siRNAs did not affect the phosphorylation (Fig. S2B). Nuclear localization of Rad9 protein and damage-induced Rad9 foci formation were not affected by the ATM specific siRNAs (Fig. S3). These results demonstrate that Rad9Ser272 becomes phosphorylated in response to DNA damage in an ATM dependent manner.
The Rad9-S272C mutant cells are defective in homologous recombination
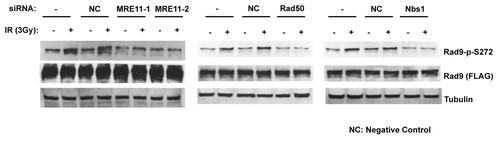
To investigate the function of Rad9Ser272 phosphorylation in controlling DNA repair pathways, we examined I-SceI-induced recombination frequencies in the Rad9-S272C and S272A mutant cells using a green fluorescent protein (GFP)-based recombination assay. This system utilizes GFP to measure recombination and the I-SceI endonuclease to introduce DSBs. The gene conversion events are readily detected by flow cytometry.Citation32 The I-SceI-induced recombination frequency was lower in the mutant cells compared with the wild type controls (p < 0.01, student’s t-test) (). This result indicates that the importance of Ser272 phosphorylation lies in controlling homologous recombination.
Figure 3. ATM-dependent Rad9 phosphorylation at Ser272 is important to control recombination pathways. The Rad9-S272C/A mutant cells show decreased homologous recombinations induced by I-SCEI, p <0.01 (student’s t-test).
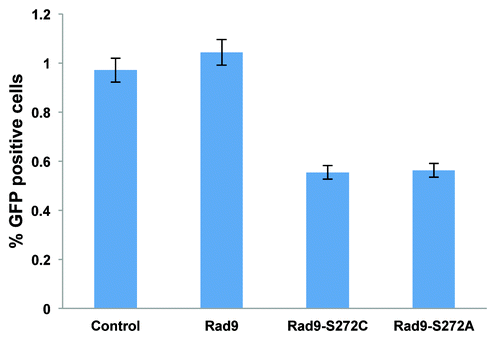
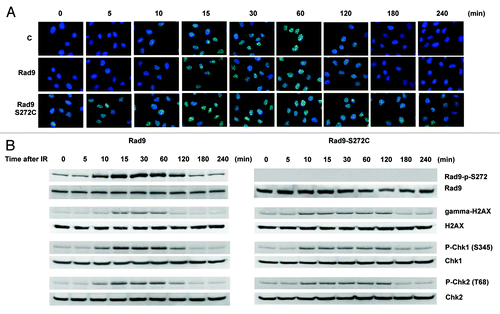
Homology-directed repair requires the Rad51-dependent homologous recombination pathway.Citation33 We therefore checked Rad51 foci formation, and found elevated levels of Rad51 foci in the mutant without DNA-damaging agents. Rad51 foci disappeared 120 min after IR treatment in wild type cells. However, the foci persisted even after 240 min in the Rad9 mutant cells, implying that Rad51-dependent recombination repair is not efficient in the mutant cells (). We also observed γ-H2AX foci during S-phase in untreated Rad9-S272C mutant cells but not in control cells, indicating that DSBs are generated in the mutant cells during unperturbed cell cycle (Fig. S6). Persistent γ-H2AX, phosphorylated Chk1 and Chk2 were detected after IR, showing delayed recovery from checkpoint activation in the mutant cells (). All these observations confirm that there are more DNA breaks, caused by defective repair control.
Figure 4. The Rad9-S272C mutant cells accumulate DNA damage. (A) Rad51 foci formation in Rad9 and Rad9-S272C cells before and after IR (3 Gy). (B) Timing of Rad9Ser272, H2AX, Chk1 and Chk2 phosphorylation in response to IR (3 Gy).
ATM-dependent Rad9Ser272 phosphorylation occurs during unperturbed cell cycle
The defect in homologous recombination repair implies that Ser272 phosphorylation might function to regulate repair pathways during cell cycle. Indeed we observed Ser272 phosphorylation throughout unperturbed S to G2 phase in synchronized culture. Unexpectedly, this S-G2 phase-dependent phosphorylation was abolished by the KU55933 treatment and ATM specific siRNAs, indicating that Rad9Ser272 became phosphorylated in an ATM-dependent manner during unperturbed cell cycle (). It is possible that the double-thymidine block to synchronize the cell cycle could cause DNA damage. However, Chk1, Chk2 and histone H2AX were not phosphorylated in this condition (). Therefore, it is unlikely that the double-thymidine block induced DSBs that activate ATM-dependent checkpoint pathways. We further confirmed ATM and MRN complex-dependent Rad9Ser272 phosphorylation in S to G2 phase with sorted cells (; Fig. S7A and S8). Rad9Ser272 phosphorylation was not affected in ATR-Seckel cells (Fig. S7B). These results demonstrate that ATM, not ATR, is the main kinase that phosphorylates Ser272, not only in response to DNA damage but during unperturbed S-G2 phase as well.
Figure 5. Rad9Ser272 is phosphorylated during normal cell cycle in an ATM dependent manner. (A) Cell cycle progression and Rad9S272 phosphorylation after double thymidine block with (right) and without KU55933 treatment (10 µM) (left). (B) Rad9Ser272 phosphorylation in sorted cells with (right) and without KU55933 (10 µM) treatment (left).
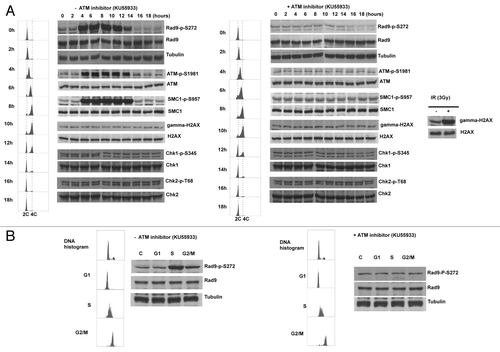
ATM is activated during unperturbed S and G2 phases
It is generally thought that ATM is activated in response to DSBs.Citation7,Citation34-Citation39 However, we observed ATM-dependent phosphorylation of Rad9 without exogenous DNA damage (; Fig. S7). Thus we examined the auto-phosphorylation status of ATM. Autophosphorylation of ATM at Ser1981 was detected during S-G2 phase in unperturbed cells. Furthermore, we observed SMC1Ser957 phosphorylation that depends on ATM.Citation40,Citation41 Interestingly, however, Chk1, Chk2 and H2AX phosphorylation was not detected in unperturbed cells (; Fig. S6 and S7). It is possible that this ATM activation can be seen only in transformed cells. Thus we investigated phosphorylation status of Rad9 and auto-phosphorylation status of ATM in a primary cell line MRC5. Both phosphorylation during S-G2 phase was observed (Fig. S9). These results demonstrate that ATM is active during unperturbed S-G2 phase of the cell cycle and phosphorylate certain substrates without a detectable level of DSBs.
We further investigated whether Rad9Ser272C mutation affects phosphorylation of other ATM substrates. We observed intact phosphorylation of ATM substrates, SMC1, NBS1, BRCA1, 53BP1 and Rad17 (Fig. S10).
The Rad9-S272C mutant cells induce chromosome instability
To assess the impact of these unscheduled repairs on chromosomal stability, we examined metaphase chromosome spreads. Fully 59% of the Rad9-S272C mutant cells exhibited more than two chromosomal breakages per cell 12 h after IR, compared with 21% of wild type Rad9 cells. A similar result was observed after CPT treatment (Rad9-S272C cells 51%, Rad9 cells 9%). The contrast was even more pronounced for spontaneous chromosomal breakages (Rad9-S272C cells 22%, Rad9 cells 1%) (). These results confirm that Rad9Ser272 phosphorylation is required for proper repair control and maintenance of replication fork stability during unperturbed cell cycle. ATM phosphorylates Rad9 in order to prevent chromosomal breakage during replication.
Figure 6. The Rad9-S272C mutant cells accumulate chromosome breakages and show chromosome instability phenotypes. (A) Chromosome breakages in Rad9 and Rad9-S272C cells. An example of metaphase spread of a Rad9-S272C cell (top, left). The Rad9-S272C cells induce a high frequency of chromosome breakages without damage (top, right), after IR treatment (3 Gy) (bottom, right), after CPT treatment (500nM, 6 h) (bottom, left). (B) The Rad9-S272C mutant cells induce a high frequency of GCRs, chromosome rearrangements. Twenty cells for each cell line were karyotyped and chromosome abnormalities were detected.
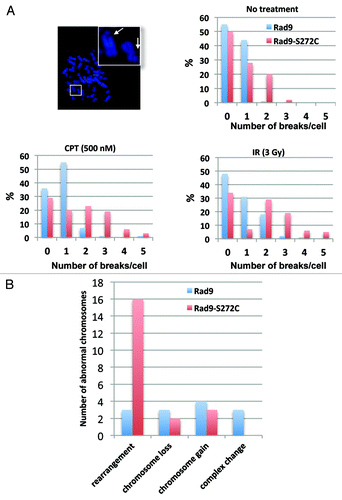
The accumulation of chromosomal breakages alone might not necessarily be disastrous, if the breaks are correctly repaired. Incorrect repairs, however, will cause deleterious long-term effects such as gross chromosomal rearrangements (GCRs). To look for these, we sought to perform karyotype analysis using human normal primary (MRC5) cells. However, the Rad9-S272C MRC5 cells were not viable, confirming the importance of phosphorylation during normal cell cycle in non-transformed cells. We therefore employed a transformed (HCT116) cell line for karyotype analysis. We did not observe significant genomic instability in p53+/+ cells (data not shown). However, the Rad9-S272C mutant cells (endogenous Rad9 knocked down) with p53−/− background showed significantly higher frequency of GCRs compared with the wild type controls (). We conclude that the improved survival of the Rad9 mutant after DNA damage comes at an unacceptable long-term cost.
Discussion
Our results demonstrate that ATM-dependent phosphorylation of Rad9 acts to maintain chromosome stability by coordinating repair pathways. This is unexpected, particularly during S to G2 phase, which is independent of Rad9-dependent checkpoint activation pathways. The absence of ATM-dependent phosphorylation induces DSBs, which are improperly repaired in the mutant. ATM detects improper DNA structures and/or replication fork collapses and signals to repair pathways through the checkpoint clamp during S to G2 phase. We created Rad9-S272C mutant cell lines that express the mutant proteins stably and the endogenous Rad9 protein is knocked down. Originally we chose Rad9-S272C mutation instead of Rad9-S272A mutation, because it has been reported that Rad9-phosphorylation site mutation on the PCNA like domain causes a more severe DNA damage sensitivity possibly due to structural defect in yeast.Citation42 We repeated experiments with Rad9-S272A mutant cells, and obtained similar results as with Rad9-S272C mutant cells. Importantly, the defects of Rad9-S272C and Rad9-S272A mutant cells in repair regulation do not reduce cell survival in transformed cells. On the contrary, the mutant cells show slightly improved survival after DNA damage (Fig. S5). The mutant cells show defect in I-SCEI induced homologous recombination. This deficiency is modest compared with other checkpoint/homologous recombination protein mutants such as BRCA1. The DSBs caused by I-SCEI are probably repaired by error-prone pathways such as non-homologous end joining and single-strand annealing. Repairs are not properly done in the mutant cells, leading to serious genomic instability. Given that the Rad9-S272C mutant cells induce deregulation of recombination and GCRs ( and ), failure of phosphorylation at this site could lead to further mutations in tumor suppressor genes. Furthermore, increased survival of the mutant cells after damage would confer a selection advantage on these cells in the event of, e.g., chemotherapy treatment. Therefore, ATM-dependent phosphorylation of the checkpoint clamp Rad9Ser272 might function to prevent progression of tumor development and malignant transformation of tumor cells.
ATM activation during unperturbed S-G2 phase of cell cycle
It is generally accepted that ATM becomes activated in response to DSBs. However, we observed autophosphorylation of ATM and phosphorylation of ATM substrates in unperturbed cells (; Fig. S8). This result is indeed consistent with the results shown in 293T cells that showed ATMSer1981 autophosphorylation without exogenous damage. Autophosphorylation was not detected in primary fibroblasts,1070S.Citation43 However, we detected phosphorylation of ATMSer1981 and ATM substrates in primary MRC5 cells that were enriched in S-G2 phase (Fig. S10). Interestingly, Chk2 and H2AX phosphorylation were not observed during unperturbed cell cycle (, Fig. S7 and S8). Therefore, ATM phosphorylates a subset of substrates during S to G2 phase of the cell cycle without “detectable” DSBs, but does not activate the checkpoint pathway. However, it is possible that ATM is activated by an undetectable level of DSBs caused by spontaneous replication fork collapse during S phase. Indeed, transient activation of ATM has been observed in Xenopus egg extracts with undamaged DNA. The authors speculate that transient generation of DSBs during replication triggers a local activation of ATM on replication chromatin. The authors also showed that ATM slows the rate of replication by inhibiting Cdk2 kinase.Citation44,Citation45 Genomic DNA replicated in Xenopus extracts immunodepleted of X-MRE11 complex accumulate DSBs. Therefore, it is thought that the function of X-MRE11 complex is to repair DSBs that arise during normal DNA replication although H2A phosphorylation was not detected in the cells. Rad9 is phosphorylated during unperturbed S to G2 phase of the cell cycle in an ATM and MRE11 dependent manner. We speculate that this phosphorylation might be required for fast repair of DSBs and/or to protect genome from replication fork collapse. Indeed, we observed elevated level of chromosome breaks in the mutant cells without exogenous damage as in the case of X-MRE11 depleted cell extracts ().
Increased spontaneous intrachromosomal as well as extrachromosomal recombinations in ATM-deficient cells have been shown by several studies.Citation46-Citation48 Cytogenetic analysis revealed a higher spontaneous incidence of chromosome breaks, chromosome gaps, acentric fragments, dicentric chromosomes and aneuploidy in ATM patient cells.Citation49,Citation50 A recent study showed that ATM could be activated without DSBs by oxidative stress. Oxidation of ATM directly induces ATM activation, and this mechanism is Mre11-Rad50-Nbs1 (MRN) complex independent.Citation51 These results demonstrate important functions of ATM to prevent genomic instability during normal cell cycle.
In this study, we discovered a new ATM-dependent pathway to protect genome stability during unperturbed cell cycle. ATM phosphorylates a subset of its substrates for proper repair control without exogenous damage. Phosphorylation of Rad9 at Ser272 by ATM promotes its dissociation from chromatin. Failure of this process induces various genomic instability phenotypes ( and ). It is yet unclear whether activation of this pathway requires spontaneous DSBs or not. Further studies are required to understand the mechanism of ATM activation during unperturbed cell cycle.
Materials and Methods
Materials, antibodies and cell lines
The Rad9-flag expressing plasmids were created by inserting Rad9 cDNA into p3xFLAG-CMV-14 (Sigma-Aldrich) and pBMN vectors. The Rad9-S272C mutation was made using QuickChange XL Site-directed Mutagenesis kit (Stratagene). Rad9 shRNA (V3LHS_401433) construct was obtainted from OpenBio Systems. The Rad9 and Rad9.S272C cell lines (stably express exogenous wild type or the mutant Rad9 proteins, and that the endogenous Rad9 expression is stably knocked down) were created first by selecting Rad9-flag expressing single clones and then infecting a shRad9 construct in the DR-293 cell line. Finally stable Rad9 knockdown cells were selected. The siATM, SI00604730 and SI00299299, siMRE11, SI02665173 and SI02665180 and negative control siRNAs were obtained from QIAGEN. The siATR, SignalSilence® ATR siRNA I (6288S), SignalSilence® ATR siRNA II (6289S) and negative control SignalSilence siRNA (6568S) were from Cell Signaling Technology. Nbs1 siRNA (h): sc-36061, Rad50 siRNA (h): sc-37397, DNA-PKCs siRNA (h): sc-35200 and Control siRNA: sc-37007 were obtained from Santa Cruz Biotechnology. Five nM siRNAs were used for the experiments.
Anti-flag antibody was from Sigma-Aldrich. Anti-Chk1, phospho-Chk1, Chk2, phospho-Chk2, ATM, phospho-ATM (S1981), SMC1, phospho-SMC1 (S957), Nbs1, phospho-Nbs1 (S343), BRCA1, phospho-BRCA1 (S1524), 53BP1, phospho-53BP1 (S25/29), Rad50, Mre11 ATR and gamma-H2AX antibodies were obtained from Cell Signaling. Anti-phospho-Rad9-S272 antibody was from Abgent. Anti-Rad51 and antibody were from Calbiochem. Anti-Rad17, phospho-Rad17 (S645) and DNA-PKCs antibodies were from abcam.
Cell synchronization and cell cycle analysis
HeLa cells were incubated with 2 mM thymidine for the first block for 18 h, and a 9 h release for the second block for 17 h, followed by a release up to 18 h. Cells were trypsinized, washed in PBS, and resuspended in 70% ethanol for storage at -20°C until further analysis. Cells were stained with propidium iodide (50 µg/ml), and analyzed with a MoFlo MLS cytometer (Beckman Coulter).
Cell survival (proliferation) assay
DR293 cells (1 x 104) cells, including control, Rad9 and Rad9-S272C cell lines were plated on 96-well plates. After 4 h incubation, cells were exposed to UV, IR, CPT and HU and incubated for 24 h. Twenty µl Cell Titer-Blue cell viability assay reagent (Promega) was added to each well, incubated for 1 h and measured with 96-well plate reader at 560Ex/590Em.
Clonogenic cell survival assay
DR293 cells including control, Rad9 and Rad9-S272C cell lines were plated (3x103 cells) on 10 cm-plate, exposed to IR, UV, CPT and HU and incubated for 14 d. The resulting colonies were stained with 0.5% Crystal violet / 50% Methanol and counted.
GFP-based recombination assay
I-SceI expression plasmids (100µg) were electroporated (250V, 959µF) into DR293 cells (5x106 cells).Citation32 After electroporation, cells were plated in non-selective media for 72 h, and recovered and concentrated by centrifugation, and resuspended in Opti-MEM media (Invitrogen) prior to flow cytometry. Cells were analyzed in FACSCalibur (BD Biosciences).
Immunofluorescence
DR293 cells, Rad9 and Rad9-S272C cell lines were fixed in 4% paraformaldehyde for 20 min, washed 3 times in PBS, and incubated for 45 min in blocking buffer containing 130 mM, NaCl, 10 mM Na2HPO4, 3 mM NaH2PO4, 1% bovine serum albumin, 2% Triton-X-100, 0.5% Tween 20 and 5% donkey serum. Incubation with primary antibody was 1 h at room temperature with the following dilutions in blocking buffer. 1:100 for Anti-Rad9 antibody (BD Biosciences), 1:200 for Anti-phospho-Rad9-S272 antibody (Abgent). Following three washes with PBS, cells were incubated for 1 h with Fluorescein (FITC) or Rhodamine (TRITC) conjugated Donkey Anti-mouse or rabbit IgG (H+L) (Jackson ImmunoResearch Laboratories) diluted to 1:100 with blocking buffer. Cells were washed three times with PBS before observation and DAPI solution was applied to stain nuclei. Cells were visualized by Zeiss imager.Z1.
Cytogenetic analysis
Colcemid (0.1 µg/ml) was added to cell culture 3 h before harvesting by tripsinazation. Cells were then swollen for 15 min at 37°C in 0.075M KCl, and fixed for 10 min in methanol:acetic acid (3:1; vol:vol). Fixed cells were spotted onto microscopic slides and stained by DAPI (4′,6-diamidino-2-phenylindole). For each sample, chromosomal breakages in at least 100 well-spread metaphase cells were counted.
Additional material
Download Zip (6.9 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank M. Jasin for providing us the DR293 cell line and the I-SceI expression plasmid. This work was supported by a K01 award (5K01CA114027) from National Institute of Health (NIH, M.K).
Note
Supplemental materials can be found at: www.landesbioscience.com/journals/cc/article/20161
References
- Kai M, Wang TS. Checkpoint responses to replication stalling: inducing tolerance and preventing mutagenesis. Mutat Res 2003; 532:59 - 73; http://dx.doi.org/10.1016/j.mrfmmm.2003.08.010; PMID: 14643429
- Kai M, Furuya K, Paderi F, Carr AM, Wang TS. Rad3-dependent phosphorylation of the checkpoint clamp regulates repair-pathway choice. Nat Cell Biol 2007; 9:691 - 7; http://dx.doi.org/10.1038/ncb1600; PMID: 17515930
- Kerzendorfer C, O’Driscoll M. Human DNA damage response and repair deficiency syndromes: linking genomic instability and cell cycle checkpoint proficiency. DNA Repair (Amst) 2009; 8:1139 - 52; http://dx.doi.org/10.1016/j.dnarep.2009.04.018; PMID: 19473885
- Massagué J. G1 cell-cycle control and cancer. Nature 2004; 432:298 - 306; http://dx.doi.org/10.1038/nature03094; PMID: 15549091
- Hartwell LH, Kastan MB. Cell cycle control and cancer. Science 1994; 266:1821 - 8; http://dx.doi.org/10.1126/science.7997877; PMID: 7997877
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432:316 - 23; http://dx.doi.org/10.1038/nature03097; PMID: 15549093
- Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol 2005; 70:99 - 109; http://dx.doi.org/10.1101/sqb.2005.70.002; PMID: 16869743
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15:2177 - 96; http://dx.doi.org/10.1101/gad.914401; PMID: 11544175
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 2004; 118:699 - 713; http://dx.doi.org/10.1016/j.cell.2004.08.015; PMID: 15369670
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003; 22:5612 - 21; http://dx.doi.org/10.1093/emboj/cdg541; PMID: 14532133
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J 2003; 22:6610 - 20; http://dx.doi.org/10.1093/emboj/cdg630; PMID: 14657032
- Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004; 304:93 - 6; http://dx.doi.org/10.1126/science.1091496; PMID: 15064416
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308:551 - 4; http://dx.doi.org/10.1126/science.1108297; PMID: 15790808
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37 - 45; http://dx.doi.org/10.1038/ncb1337; PMID: 16327781
- Helt CE, Wang W, Keng PC, Bambara RA. Evidence that DNA damage detection machinery participates in DNA repair. Cell Cycle 2005; 4:529 - 32; http://dx.doi.org/10.4161/cc.4.4.1598; PMID: 15876866
- Yoo HY, Shevchenko A, Shevchenko A, Dunphy WG. Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J Biol Chem 2004; 279:53353 - 64; http://dx.doi.org/10.1074/jbc.M408026200; PMID: 15448142
- Gatei M, Zhou BB, Hobson K, Scott S, Young D, Khanna KK. Ataxia telangiectasia mutated (ATM) kinase and ATM and Rad3 related kinase mediate phosphorylation of Brca1 at distinct and overlapping sites. In vivo assessment using phospho-specific antibodies. J Biol Chem 2001; 276:17276 - 80; http://dx.doi.org/10.1074/jbc.M011681200; PMID: 11278964
- Chen MJ, Lin YT, Lieberman HB, Chen G, Lee EY. ATM-dependent phosphorylation of human Rad9 is required for ionizing radiation-induced checkpoint activation. J Biol Chem 2001; 276:16580 - 6; http://dx.doi.org/10.1074/jbc.M008871200; PMID: 11278446
- Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 2002; 16:571 - 82; http://dx.doi.org/10.1101/gad.970702; PMID: 11877377
- Yuan SS, Chang HL, Hou MF, Chan TF, Kao YH, Wu YC, et al. Neocarzinostatin induces Mre11 phosphorylation and focus formation through an ATM- and NBS1-dependent mechanism. Toxicology 2002; 177:123 - 30; http://dx.doi.org/10.1016/S0300-483X(02)00220-2; PMID: 12135616
- Zhang X, Succi J, Feng Z, Prithivirajsingh S, Story MD, Legerski RJ. Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol Cell Biol 2004; 24:9207 - 20; http://dx.doi.org/10.1128/MCB.24.20.9207-9220.2004; PMID: 15456891
- Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci 2009; 122:4070 - 80; http://dx.doi.org/10.1242/jcs.053702; PMID: 19843584
- Roos-Mattjus P, Hopkins KM, Oestreich AJ, Vroman BT, Johnson KL, Naylor S, et al. Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J Biol Chem 2003; 278:24428 - 37; http://dx.doi.org/10.1074/jbc.M301544200; PMID: 12709442
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics 2010; 9:1314 - 23; http://dx.doi.org/10.1074/mcp.M900616-MCP200; PMID: 20164059
- Bao S, Lu T, Wang X, Zheng H, Wang LE, Wei Q, et al. Disruption of the Rad9/Rad1/Hus1 (9-1-1) complex leads to checkpoint signaling and replication defects. Oncogene 2004; 23:5586 - 93; http://dx.doi.org/10.1038/sj.onc.1207753; PMID: 15184880
- Weiss RS, Enoch T, Leder P. Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev 2000; 14:1886 - 98; PMID: 10921903
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-04-2727; PMID: 15604286
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21:4129 - 39; http://dx.doi.org/10.1128/MCB.21.13.4129-4139.2001; PMID: 11390642
- Ward IM, Minn K, Chen J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 2004; 279:9677 - 80; http://dx.doi.org/10.1074/jbc.C300554200; PMID: 14742437
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 2003; 33:497 - 501; http://dx.doi.org/10.1038/ng1129; PMID: 12640452
- Wilsker D, Bunz F. Loss of ataxia telangiectasia mutated- and Rad3-related function potentiates the effects of chemotherapeutic drugs on cancer cell survival. Mol Cancer Ther 2007; 6:1406 - 13; http://dx.doi.org/10.1158/1535-7163.MCT-06-0679; PMID: 17431119
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13:2633 - 8; http://dx.doi.org/10.1101/gad.13.20.2633; PMID: 10541549
- Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 1999; 401:397 - 9; http://dx.doi.org/10.1038/43932; PMID: 10517641
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108:73 - 112; http://dx.doi.org/10.1016/B978-0-12-380888-2.00003-0; PMID: 21034966
- Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett 2010; 584:3675 - 81; http://dx.doi.org/10.1016/j.febslet.2010.05.031; PMID: 20580718
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007; 26:7741 - 8; http://dx.doi.org/10.1038/sj.onc.1210872; PMID: 18066086
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle 2007; 6:931 - 42; http://dx.doi.org/10.4161/cc.6.8.4180; PMID: 17457059
- Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004; 3:889 - 900; http://dx.doi.org/10.1016/j.dnarep.2004.03.029; PMID: 15279774
- Rotman G, Shiloh Y. ATM: a mediator of multiple responses to genotoxic stress. Oncogene 1999; 18:6135 - 44; http://dx.doi.org/10.1038/sj.onc.1203124; PMID: 10557105
- Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev 2004; 18:1423 - 38; http://dx.doi.org/10.1101/gad.1200304; PMID: 15175241
- Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev 2002; 16:560 - 70; http://dx.doi.org/10.1101/gad.970602; PMID: 11877376
- Furuya K, Miyabe I, Tsutsui Y, Paderi F, Kakusho N, Masai H, et al. DDK phosphorylates checkpoint clamp component Rad9 and promotes its release from damaged chromatin. Mol Cell 2010; 40:606 - 18; http://dx.doi.org/10.1016/j.molcel.2010.10.026; PMID: 21095590
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499 - 506; http://dx.doi.org/10.1038/nature01368; PMID: 12556884
- Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 2004; 6:648 - 55; http://dx.doi.org/10.1038/ncb1145; PMID: 15220931
- Costanzo V, Robertson K, Bibikova M, Kim E, Grieco D, Gottesman M, et al. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell 2001; 8:137 - 47; http://dx.doi.org/10.1016/S1097-2765(01)00294-5; PMID: 11511367
- Meyn MS. High spontaneous intrachromosomal recombination rates in ataxia-telangiectasia. Science 1993; 260:1327 - 30; http://dx.doi.org/10.1126/science.8493577; PMID: 8493577
- Bishop AJ, Barlow C, Wynshaw-Boris AJ, Schiestl RH. Atm deficiency causes an increased frequency of intrachromosomal homologous recombination in mice. Cancer Res 2000; 60:395 - 9; PMID: 10667593
- Drexler GA, Wilde S, Beisker W, Ellwart J, Eckardt-Schupp F, Fritz E. The rate of extrachromosomal homologous recombination within a novel reporter plasmid is elevated in cells lacking functional ATM protein. DNA Repair (Amst) 2004; 3:1345 - 53; http://dx.doi.org/10.1016/j.dnarep.2004.05.001; PMID: 15336629
- Kojis TL, Gatti RA, Sparkes RS. The cytogenetics of ataxia telangiectasia. Cancer Genet Cytogenet 1991; 56:143 - 56; http://dx.doi.org/10.1016/0165-4608(91)90164-P; PMID: 1756458
- Cohen MM, Levy HP. Chromosome instability syndromes. Adv Hum Genet 1989; 18:43 - 149, 365-71; http://dx.doi.org/10.1007/978-1-4613-0785-3_2; PMID: 2658496
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330:517 - 21; http://dx.doi.org/10.1126/science.1192912; PMID: 20966255