Abstract
Cisplatin chemoresistance is a clinical problem that leads to treatment failure in various human epithelial cancers. Members of tumor protein (TP) p53 family play various critical roles in the multiple molecular mechanisms underlying the chemoresistance of tumor cells. However, the in-depth mechanisms of the cellular response to cisplatin-induced cell death are still under thorough investigation. We previously showed that squamous cell carcinoma (SCC) cells exposed to cisplatin display an ATM-dependent phosphorylation of ΔNp63α, leading to a specific function of the phosphorylated (p)-ΔNp63α transcription factor in cisplatin-sensitive tumor cells. We further found that SCC cells expressing non-p-ΔNp63α-S385G became cisplatin-resistant. Using quantitative mass-spectrometry of protein complexes labeled with isobaric tags, we showed that TP53 and ΔNp63α are involved in numerous protein-protein interactions, which are likely to be implicated in the response of tumor cells to cisplatin exposure. We found that p-ΔNp63α binds to the splicing complex, leading to repression of mRNA splicing and activation of ACIN1-mediated cell death pathway. In contrast to p-ΔNp63α, non-p-ΔNp63α fails to bind the critical members of the splicing complex, thereby leading to activation of RNA splicing and reduction of cell death pathway. Overall, our studies provide an integrated proteomic platform in making a case for the role of the p53/p63 interactome in cisplatin chemoresistance.
Introduction
Cisplatin is the most favorable chemotherapeutic agent for treating human cancers, because it induces apoptosis of tumor cells.Citation1 However, some of the tumor cells became cisplatin-resistant during the course of chemotherapy.Citation1-Citation3 After cisplatin exposure, cells are often undergoing DNA damage leading to an accumulation of activated members of the tumor protein (TP) p53 family, which, in turn, alter the expression of a large set of the downstream target genes leading to cell cycle arrest, apoptosis, autophagy, cellular senescence and increased DNA repair.Citation4,Citation5 We previously found that squamous cell carcinoma (SCC) cells exposed to cisplatin displayed the ATM-dependent phosphorylation of wild type (wt)-ΔNp63α generating phosphorylated (p)-ΔNp63α, which is critical for the transcriptional regulation of downstream mRNAs and microRNAs and is likely to underlie the chemosensitivity of SCC cells.Citation6,Citation7 However, SCC cells expressing non-p-ΔNp63α-S385G became cisplatin resistant.Citation7 Understanding the multiple mechanisms by which (p)-ΔNp63α along with its protein interactors modulates the response of SCC cells to cisplatin will provide us with a crucial information about a role for the ATM-dependent ΔNp63α pathway in the resistance of tumor cells to platinum therapy.
The quest for novel TP63-interacting proteins is always our research priority, since it allows us to further define the critical role of TP63 in cancer, cell proliferation or apoptosis. Modern technologies emerging recently provide us with new opportunities, as well as challenges. So far only Tp53 and TP73-interacting proteins were systematically analyzed and cataloged.Citation8,Citation9 However, the global analysis of TP63 interactome is still underway. Although Tp53 family proteins (TP53, TP63 and TP73) may share many of their interactors, both TP63 and TP73 could display similar or different effects. While TP53, TP63 and TP73 were asserted forming homodimers, TP63 isoforms were found to interact with Tp53 using their mutual DNA binding domains.Citation8,Citation10 A few proteins were reported to bind the TP63 DNA binding domain or C terminus of the longest TP63 isotype (p63α) using two-hybrid system and immunoprecipitation.Citation10-Citation15
A few studies of ΔNp63α protein interactors have been reported, beginning with the first report of a Tp53 protein interacting with ΔNp63α identified by our group.Citation10 Other ΔNp63α protein interactors include members of the Wnt-signaling pathway, various transcription factors, the RNA processing complex, proteasome-dependent protein machinery and degradation machinery and IKKB.Citation12-Citation18 Using high-throughput affinity pull-downs followed by mass spectrometry (MS), Viola Calabro’s research team has elegantly showed a few novel ΔNp63-interacting proteins involved in RNA processing and splicing among other 49 interacting candidates in human lung cancer cells.Citation19
Although both Tp53 and ΔNp63α are involved in multiple protein-protein interactions, none of the studies were designed to quantify the changes in TP53/ΔNp63α interactome upon cisplatin exposure. The yeast two-hybrid screens and affinity pull-downs followed by MS do not allow such quantification. Furthermore, none of the prior studies explored the role for phosphorylation of ΔNp63α in the specific TP53- and ΔNp63α-protein interactions or examined the drug-mediated changes in the TP53-/ ΔNp63α-mediated protein interactions.
We attempted to fill this gap and to study the global TP53/TP63 interactome using a protein array chip analysis.Citation20 We assessed changes in TP53 and ΔNp63α protein-protein interactions affected by cisplatin exposure of sensitive and resistant SCC cells using the “isobaric tags for relative and absolute quantification” (iTRAQ) method.Citation21 This is the first systematic study to identify and quantify differential interactions of TP53, p-ΔNp63α and non-p-ΔNp63α in SCC cells. Potential outcome of the follow-up studies could generate novel knowledge about the roles of Tp53 and TP63 protein interactions in SCC’s biological functions and are to likely furnish new targets implicated in tumor cell viability and chemoresistance.
Results
Identification of global Tp53- and TP63-interacting proteins by protein microarray chip.
To identify the global Tp53 and TP63 interacting partners, we used the protein binding microarray approach, allowing the high-throughput screening of the protein interacting candidates among the 16,368 individual human GST-His6-tagged proteins spotted on the chip.Citation20,Citation22-Citation24 We used 25 µg of recombinant glutathione-S-transferase (GST)-Tp53 (1–393 amino acid residues) and GST-TP63 (1–680 amino acid residues) fusions purified from the baculovirus-infected Sf9 insect cells and labeled with Alexa-Flour-647. After each screen that was performed at the stringent conditions in the presence of 5–10 mM DTT, the fluorescence intensity of the spots was normalized against background, and the reproducibility of the duplicate spots was assessed. Signals less than 10% of the maximal signal were considered as background noise. The distribution of the fluorescence intensity, which reflected the binding of the bait protein with chip-embedded preys, was plotted (data not shown) to determine if the signals in the peak of the curve were essentially selected and were found suitable for subsequent bioinformatics analysis. Resulting fluorescence intensity for the Tp53 chip array analysis ranged from 13,301 to 1,300, while the intensity for the TP63 chip array analysis ranged from 24,113 to 2,400 (data not shown).
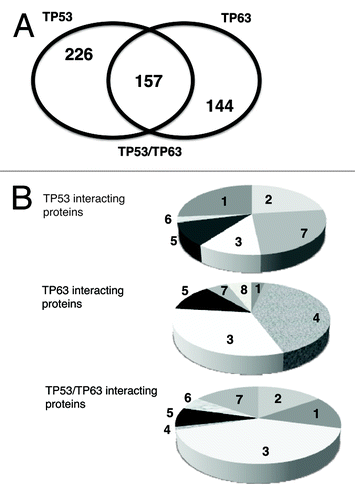
We found that Tp53 bound to 383 individual proteins (Tables S1 and S3), while TP63 bound to 301 proteins (Tables S2 and S3). All potential Tp53- and TP63-interacting partners were organized into the following groups: TP53-specific, TP63‑specific and TP53/TP63 common protein interactors (Tables S1–S3, respectively). A Venn diagram () depicts the number of proteins exclusively bound to Tp53 (226), or to TP63 (144) or both (157). Based on potential functions defined by the Gene Ontology (GO) software, TP53-specific, TP63-specific and TP53/TP63 common interacting proteins fall into the eight following categories: (1) cell death/survival; (2) DNA damage/signaling; (3) chromatin remodeling/gene regulation; (4) RNA processing; (5) protein trafficking/degradation; (6) oncogenes; (7) other and (8) epithelial differentiation (). TP53-specific interacting proteins were associated with cell death/survival, DNA damage/signaling, chromatin remodeling/gene regulation, protein trafficking/degradation, oncogenes and other (, upper part). We categorized the TP63-specific interactors as implicated in cell death/survival, chromatin remodeling/gene regulation, RNA processing, protein trafficking/degradation, other and epithelial differentiation (, middle part). However, TP53/TP63 common interactors were associated with cell death/survival, DNA damage/signaling, chromatin remodeling/gene regulation, RNA processing, protein trafficking/degradation, oncogenes and other (, lower part).
Figure 1. Bioinformatics of TP53 and TP63 interacting proteins. (A) Using the GO software the TP53/TP63 interacting partners defined by protein array chip were organized as TP53-specific, TP53/TP63 common and TP63-specific protein interactors. Venn diagram depicts a number of proteins bound to TP53, TP63 or both. (B) TP53-specific, TP53/TP63 common and TP63-specific interacting proteins were divided into eight categories representing the following functions: (1) cell death/survival; (2) DNA damage/signaling; (3) chromatin remodeling/gene regulation; (4) RNA processing; (5) protein trafficking/degradation; (6) oncogenes; (7) other and (8) epithelial differentiation.
Using Ingenuity Pathway Analysis (IPA) software, we further examined the role for the global Tp53 and TP63 protein interactors in cellular functions, pathways and networks. Among other functions, both Tp53 and TP63 were found to be involved in the following critical cellular functions: gene expression, cell cycle, cell death, cell growth/proliferation, cellular development, DNA replication, recombination and repair, cancer, developmental/dermatological disorders, embryonic development, posttranslational modifications and cellular response to therapeutics (Fig. S1, upper part). Both Tp53 and TP63 were further found to be involved in key signaling pathways: Tp53 signaling, cell cycle regulation, molecular mechanisms of cancer, G1/S checkpoint regulation, ATM signaling, G2/M checkpoint regulation, CHK signaling, epigenetic regulation, PI3K/AKT signaling, apoptosis signaling, NFκB signaling and mTOR signaling (Fig. S1, lower part). Various networks were found to involve TP53, TP63 or both: cell cycle/cell death; DNA replication/recombination/repair/cell cycle; gene expression; epigenetic regulation and RNA post-transcriptional regulation and modification, as depicted by the STRING 9.0 protein interaction database (Fig. S2).
Quantitative assessment of Tp53 and ΔNp63α protein interacting candidates by iTRAQ.
We further examined the quantitative changes in the Tp53 and ΔNp63α-interactome in SCC cells under native conditions using the iTRAQ technology.Citation25-Citation29 TP53- and ΔNp63α-specific protein complexes were purified by the Interplay N-terminal tandem affinity purification (NTAP) system. The wt-ΔNp63α, ΔNp63α-S385G and Tp53 cDNA sequences were fused to the N-terminal calmodulin (CaBP) and streptavidin (SBP) double tags. The NTAP-ΔNp63α and NTAP-Tp53 constructs were introduced into wt-ΔNp63α cells, while the NTAP-ΔNp63α-S385G and NTAP-Tp53 constructs were transfected in ΔNp63α-S385G cells along with an empty vector. Resulting cells were next exposed to 10 µg/ml of cisplatin for 16 h. NTAP proteins were purified from total lysates, stained with silver and blotted with antibodies to ΔNp63, to p-ΔNp63 and to TP53. We found that both endogenous and exogenous ΔNp63α-wt was recognized by anti-p-ΔNp63α antibody, while endogenous and exogenous ΔNp63α-S385G was not recognized by this antibody (data not shown).
We have first assessed what proteins interact with Tp53 or ΔNp63α in SCC cells transfected with the NTAP-Tp53 or NTAP-ΔNp63α expression constructs but exposed to control medium only. NTAP precipitates containing Tp53 or ΔNp63α and their respective interacting proteins were eluted from the TAP resins (first from SBP-resin and then from CaBP-resin). TP53- and ΔNp63α-specific precipitates digested with trypsin were subjected to subsequent iTRAQ labeling followed by liquid chromatography and tandem mass spectrometry (LS-MS/MS) protein identification analysis.Citation26,Citation28,Citation29 After removing the proteins that failed to score above background (of note, the iTRAQ ratios were normalized by total protein and only proteins identified with median ratios > 1.2 or < 0.8 were considered), we identified 444 total NTAP-TP53 and 310 total NTAP-ΔNp63α interactors (data not shown). The majority of the significantly enriched proteins appeared to be involved in RNA transcription (DDIT3, Tp53 and TP63), processing (SF3A2, SAP18 and HNRNPAB) and chromatin remodeling (EZH2, KAT5 and HDAC6) as shown on a “Volcano” plot (Fig. S3, upper left and right quarters). We observed that there is a fair correlation (~35– 40%) between protein interactors obtained by protein array chip analysis (383 for Tp53 and 301 for TP63) and iTRAQ analysis (444 for TP53, 310 for ΔNp63α), which could be potentially explained by the nature of the endogenous protein interactions (e.g., direct vs. indirect, need for posttranslational modifications, association with cellular compartments, low protein abundance).
We next used total lysates from wt-ΔNp63α cells expressing NTAP-ΔNp63α and NTAP-Tp53 as well as from ΔNp63α-S385G cells expressing NTAP-ΔNp63α-S385G and NTAP-TP53. All resulting cell lines were exposed to control media (two samples) and 10 µg/ml of cisplatin (four samples) for 16 h. We quantitatively compared the TP53- or ΔNp63α-protein complexes in wt-ΔNp63α cells or ΔNp63α-S385G cells exposed to control medium or cisplatin. Since cisplatin was shown to induce the ATM-dependent phosphorylation of ΔNp63α,Citation6 the ΔNp63α complexes in wt-ΔNp63α cells essentially contained p-ΔNp63α, while ΔNp63α-S385G cell-protein complexes contained non-p-ΔNp63α. NTAP protein complexes were precipitated, and the trypsin-digested peptides of the sample were labeled with isobaric tags 113 and 114 (for samples obtained from cells exposed to control media) and 115–118 (for samples obtained from cells exposed to cisplatin).
We finally identified ~197 proteins in sensitive wt-ΔNp63α cells and 169 in resistant ΔNp63α-S385G cells whose interactions to Tp53 or ΔNp63α were enriched or depleted upon cisplatin exposure (shown by two or more unique MS/MS spectra). The iTRAQ ratios ranged from 0.206 (depleted) to 4.139 (enriched) for Tp53 and 0.129 (depleted) to 3.286 (enriched) for ΔNp63α (Tables S4–S7). We observed that Tp53 displayed 83 enriched interactions and 43 depleted interactions in sensitive wt-ΔNp63α cells upon cisplatin exposure (Table S4). However, in resistant ΔNp63α-S385G cells, Tp53 displayed 69 enriched interactions but only 50 depleted interactions (Table S5). Since, ΔNp63α in sensitive wt-ΔNp63α cells is, in fact, p-ΔNp63α, the enriched interactions of the latter were associated with 48 proteins, while depleted interactions were associated with 41 proteins. On the other hand, non-p-ΔNp63α in resistant ΔNp63α-S385G cells displayed 33 enriched interactions and 37 depleted interactions (Tables S4 and S7).
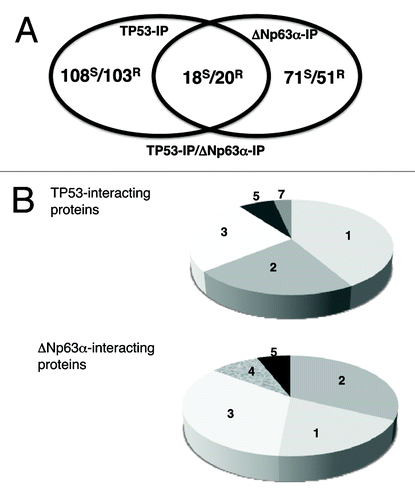
After GO analysis, interacting candidates were divided into the following groups: TP53-specific (108 from sensitive cells, 99 from resistant cells), ΔNp63α-specific (71 from sensitive cells, 50 from resistant cells) and TP53/ΔNp63α common proteins (18 from sensitive cells, 20 from resistant cells) as shown (). Furthermore, TP53-interacting proteins were categorized in the following functions: cell death/survival, DNA damage/signaling, chromatin remodeling/gene regulation, protein trafficking/degradation and other (, upper part), whereas ΔNp63α interactors were categorized in the following functions: cell death/survival, DNA damage/signaling, chromatin remodeling/gene regulation, RNA processing and protein trafficking/degradation (, lower part).
Figure 2. Bioinformatics of TP53 and ΔNp63α interacting proteins. (A) Using the GO software the TP53-IP/ΔNp63α-IP interacting partners defined by iTRAQ were organized as TP53-IP-specific, ΔNp63α-IP-specific, and TP53-IP/ΔNp63α -IP common protein interactors. Venn diagram depicts a number of proteins bound to TP53, ΔNp63α or both. S, sensitive cells; R, resistant cells. (B) TP53-specific and ΔNp63α-specific interacting proteins were divided into seven categories representing the following functions: (1) cell death/survival; (2) DNA damage/ signaling; (3) chromatin remodeling/gene regulation; (4) RNA processing; (5) protein trafficking/degradation; (6) oncogenes and (7) other.
Using the IPA program, we further evaluated a potential biological relevance of differential interactors and categorized them as cellular functions and pathways (Figs. S4 and S5). We found that both TP53- and p-ΔNp63α-protein interactions in sensitive wt-ΔNp63α cells exposed to cisplatin were enriched in proteins more related to cell death (with fewer related to cell survival). Whereas TP53- and especially non-p-ΔNp63α-protein complexes in resistant ΔNp63α-S385G cells exposed to cisplatin were enriched in proteins more related to cell survival (with fewer related to cell death). We also found that protein complexes for Tp53 and ΔNp63α were enriched in proteins related to chromatin remodeling and modifications, RNA transcription and RNA processing and proteasome-degradation machinery. Intriguingly, while most of the protein interactions involving p-ΔNp63α (sensitive cells) and non-p-ΔNp63α (resistant cells) were found to be common, some interactions were enriched with p-ΔNp63α and diminished with non-p-ΔNp63α (e.g., SAP18, E2F1, SRSF2, ACIN1, NF-YB, HDAC6, SIN3A, DDIT3 and SF3A2) and some displayed an opposite tendency (e.g., HDAC1 and -2, SIRT1, SREBF1, NAC1, CITED, HMGB1 and BECN1), as shown in Tables S4 and S7. Both Tp53 and ΔNp63α shared several protein interactions that are common between these baits (). We therefore selected a few of these examples (ACIN1, SAP18 and HDAC6) to further pursue the investigation of the TP53/ΔNp63α protein interactions as potential mechanisms for cisplatin chemoresistance.
Table 1. Overlapping cisplatin (CIS)-induced TP53- and ΔNp63- protein interactions in sensitive/resistant cells
P-ΔNp63α and non-p-ΔNp63α play opposing roles in regulation of RNA splicing and cell death response of SCC cells to cisplatin.
Accumulating evidence has strengthened the potential connection between RNA processing and cell death processes implicated in cell response to chemotherapy. Recent reports show that numerous apoptotic factors are regulated through an alternative splicing mechanism, resulting in production of discrete isoforms with distinct, if not antagonistic, functions (e.g., FAS, TRAF, APAF1, SMAC/Diablo, BCL-x, BAK, BAD, BIM, CASP2, -9 and -10, ICAD, ACIN1, TP53, TP63 and TP73, as reviewed in ref. Citation30).
Regulation of splicing can be achieved by reversible phosphorylation of the serine (S)/arginine (R) splicing regulatory (SR) proteins and ACIN1 by SR protein kinases (SRPK), which was shown to be altered during apoptosis, potentially leading to reorganization of the spliceosome.Citation31,Citation32 Although, the nuclear protein ACIN1 is known to function in DNA fragmentation and is activated during apoptosis by CASP3, it plays an active role in spliceosome assembly, as it is a critical subunit of an apoptosis and splicing-associated protein (ASAP) complex that consists of SAP18 and the splicing activator RNPS1 (as reviewed in refs. Citation33 and Citation34). ASAP complex formation was shown to represses the RNA processing mediated by SRSF1 and -2, or by RNPS1; however, ASAP microinjection into mammalian cells resulted in acceleration of cell death.Citation33 Splicing regulatory proteins, SRSF1 and -2, and RBM5 were shown to promote exon 9 skipping in Casp2, causing an increase of proapoptotic CASP2L, while HNRNPA1 reduced exon 9 skipping, leading to an increase of anti-apoptotic CASP2S (reviewed in refs. Citation35 and Citation36).
We previously found that the wt-ΔNp63α protein is tightly implicated in the RNA processing mechanism by binding to HNRNPAB and SCAF4, while naturally derived mutations in the p63α C-terminal region associated with ectodermal dysplasia dramatically abolished this interaction.Citation13,Citation37 We, thus, further examined the role for p-ΔNp63α or non-p-ΔNp63α in protein-protein interactions involved in RNA splicing, which were modulated by exposure of SCC cells to cisplatin.
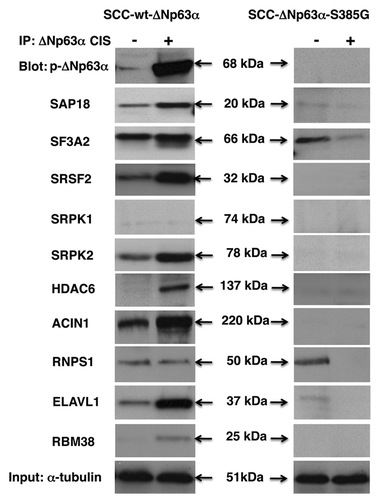
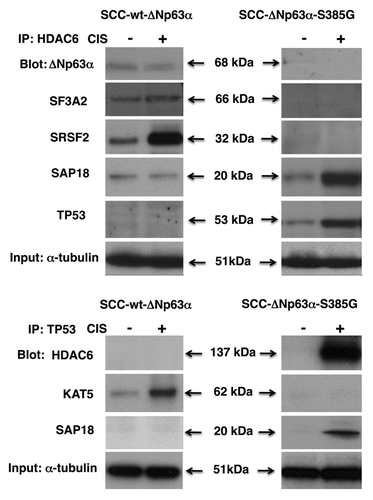
We obtained nuclear lysates from sensitive wt-ΔNp63α cells or resistant ΔNp63α-S385G cells exposed to cisplatin treatment leading to phosphorylation of the former (p-ΔNp63α), while the latter remained unchanged (non-p-ΔNp63α). We show here that the cisplatin exposure induced the ΔNp63α (notably p-ΔNp63α) protein complexes with SAP18, SF3A2, SRSF2, SRPK2, HDAC6 and ACIN1 in sensitive cells (, left parts), while no interaction of non-p-ΔNp63α was shown with SAP18, SRSF2, SRPK2 or ACIN1, and levels of protein interaction with SF3A2 dramatically decreased in resistant cells upon cisplatin exposure (, right parts). Additionally, we found that p-ΔNp63α associated with proteins that regulate mRNA stability, including ELAVL1 and RBM38 (, left parts), while non-p-ΔNp63α failed to bind both ELAVL1 and RBM38 (, right parts). We also observed that cisplatin exposure did not change the level of interaction between p-ΔNp63α and RNPS1 (, left parts), while it dramatically reduced the binding of non-p-ΔNp63α to RNPS1 (, right parts). No interaction with SRPK1 was found in either of the experimental conditions ().
Figure 3. Complex formation between ΔNp63α and splicing protein complex in SCC cells upon cisplatin exposure. Wt-ΔNp63α cells (left panels) and ΔNp63α -S385G cells (right panels) were exposed to control medium (CIS, -) or 10 μg/ml cisplatin (CIS, +) for 16 h. Nuclear lysates were immunoprecipitated (IP) with an anti-ΔNp63 antibody and blotted with indicated antibodies. Inputs (10%) were tested with anti-α-tubulin.
We further observed that the cisplatin treatment of sensitive wt-ΔNp63α cells led to the binding of SAP18 to ΔNp63α, SRSF2, SRPK2, ACIN1, RNPS1 (, left parts), while in resistant SCC cells, protein association of SAP18 was shown with both HDAC6 and Tp53 (, right parts). SAP18 was shown to form complexes with SF3A2 in both experimental conditions (). We further found that HDAC6 bound to SF3A2, SRSF2 and slightly to SAP18 and ΔNp63α in sensitive wt-ΔNp63α cells (, left parts), while in resistant ΔNp63α-S385G cells, HDAC6 formed a substantial amount of protein complexes with both SAP18 and TP53, while the ability of HDAC6 to bind SRSF2 was abolished (, right parts). On the other hand, Tp53 associated with KAT5 in sensitive wt-ΔNp63α cells but did not bind to either HDAC6 or SAP18 (, left parts). Alternatively, Tp53 was found to associate with both HDAC6 and SAP18, while it failed to bind KAT5 in resistant ΔNp63α-S385G cells (, right parts).
Figure 4. Complex formation between splicing protein complex and ΔNp63α in SCC cells upon cisplatin exposure. Wt-ΔNp63α cells (left panels) and ΔNp63α -S385G cells (right panels) were exposed to control medium (CIS, -) or 10 μg/ml cisplatin (CIS, +) for 16 h. Nuclear lysates were immunoprecipitated (IP) with an anti-SAP18 antibody and blotted with indicated antibodies. Inputs (10%) were tested with anti-α-tubulin.
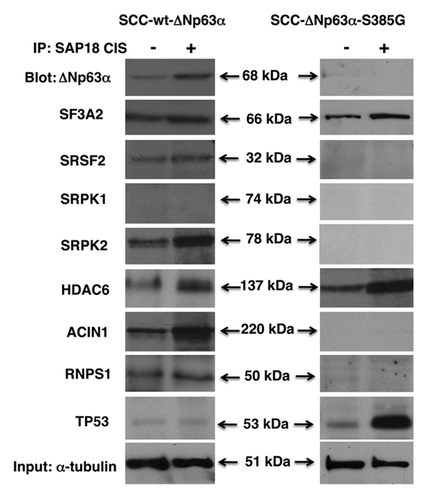
Figure 5. Complex formation between HDAC6, TP53 and splicing complex in SCC cells upon cisplatin exposure. Wt-ΔNp63α cells (left panels) and ΔNp63α -S385G cells (right panels) were exposed to control medium (CIS, -) or 10μg/ml cisplatin (CIS, +) for 16 h. (A) Nuclear lysates were immunoprecipitated (IP) with an anti-HDAC6 antibody and blotted with indicated antibodies. (B) Nuclear lysates were immunoprecipitated (IP) with an anti-TP53 antibody and blotted with indicated antibodies. Inputs (10%) were tested with anti-α-tubulin.
A recent report shows that splicing factors (e.g., Ski-interacting protein SKIP/SNW1, SRSF2) function as regulators of transcription and splicing of CDKN1A.Citation38,Citation39 Moreover, SKIP depletion induces a rapid downregulation of CDKN1A, thereby predisposing cells to undergo p53-mediated apoptosis greatly enhanced by chemotherapeutic agents.Citation39 To further examine the significance of the ΔNp63α in splicing/apoptosis regulation, we transfected wt-ΔNp63α cells with an empty vector or ΔNp63α-S385G-FL expression cassette, while ΔNp63α-S385G cells were transfected with an empty vector or ΔNp63α-FL. Resulting cells were exposed to control media or 10 µg/ml cisplatin for 16 h. Using qPCR assay, we then tested the spliced (SP) and unspliced (US) levels of CDKN1A pre-mRNA and observed that the cisplatin exposure decreased the SP-RNA/US-RNA ratio in wt-ΔNp63α cells ( and left part), while this ratio increased in ΔNp63α-S385G cells (, right part). Interestingly, the forced expression of ΔNp63α-S385G-FL (S385G-FL) dramatically induced the SP/US ratio for CDKN1A pre-mRNA in wt-ΔNp63α cells (, left part); however, the forced expression of wt-ΔNp63α-FL (WT-FL) significantly reduced that ratio in ΔNp63α-S385G cells (, right part). We also show that the cyclin A1 (CCNA1)/cyclin A2 (CCNA2) ratio was decreased in wt-ΔNp63α cells after cisplatin treatment, while it was increased by S385G-FL expression (, left part), and was alternatively increased in ΔNp63α-S385G cells after cisplatin treatment, while decreased by the WT-FL expression (, right part).
Figure 6. Opposite effect of p-ΔNp63α and ΔNp63α-S385G on regulation of mRNA splicing and DNA fragmentation in SCC cells upon cisplatin exposure. Wt-ΔNp63α cells (left panels) were transfected with an empty vector and ΔNp63α-S385G-FL cassette (S385G-FL), while ΔNp63α-S385G cells (right panels) were transfected with an empty vector and ΔNp63α-FL cassette (WT-FL). Cells were exposed to control medium or 10 μg/ml cisplatin for 16 h. (A) Cells were tested for splicing of p21CIP1/WAF1 (CDKN1A) and expression of CCNA1 and CCNA2 isoforms. (B) Cells were tested for DNA fragmentation.
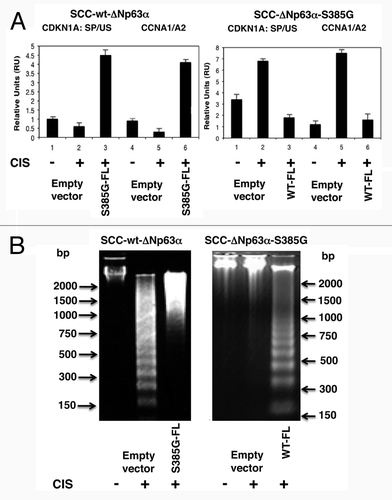
Since, ACIN1 was reported to regulate the fragmentation of nuclear DNA,Citation34 we evaluated the role of ΔNp63α-mediated protein interaction with ASAP complex in SCC cells exposed to cisplatin treatment. We show that the cisplatin exposure induced DNA fragmentation in wt-ΔNp63α cells, and forced expression of S385G-FL reduced that effect (, left part); however, no DNA fragmentation was found in ΔNp63α -S385G cells upon cisplatin exposure unless the WT-FL expression construct was added (, right part).
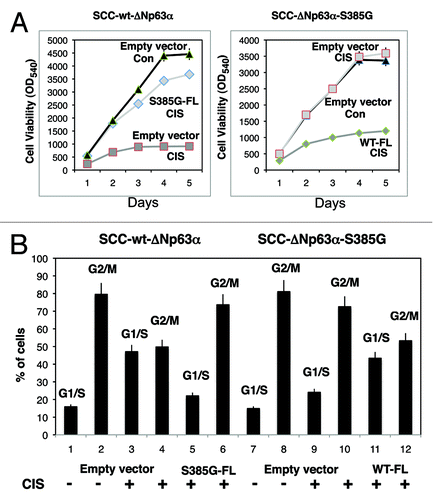
With the aid of MTT cell viability assay, we show that wt-ΔNp63α cells transfected with an empty vector substantially decreased their survival, while the forced expression of S385G-FL partially restored the cell viability under cisplatin treatment (, left part). However, ΔNp63α-S385G cells transfected with an empty vector failed to be affected by cisplatin exposure, while the forced expression of WT-FL dramatically decreased the cell viability under cispaltin treatment (, right part). Using flow cytometry analysis, we further show that the cisplatin exposure led to a marked increase in G1/S cell population and a corresponding decrease of G2/M cells at 48 h after transfection of wt-ΔNp63α cells with an empty vector, while cells transfected with the S385G-FL expression cassette show no significant change compared with cells treated with control medium (, left part, samples 1–6). However, when ΔNp63α-S385G cells were transfected with an empty vector, the cisplatin exposure minimally changed the G1/S and G2/M cell populations compared with cells treated with control media, while the forced expression of the WT-FL cassette substantially increased G1/S cell population, as G2/M cell population decreased (, right part, samples 7–12). These data suggest that p-ΔNp63α can activate cell cycle arrest, while non-p-ΔNp63α could inhibit cell cycle arrest under these experimental conditions.
Figure 7. Opposite effect of p-ΔNp63α and ΔNp63α-S385G on cell viability and cell cycle arrest of SCC cells upon cisplatin exposure. Wt-ΔNp63α cells (left panels) were transfected with an empty vector (triangles and squares) and ΔNp63α-S385G-FL cassette (S385G-FL, diamonds), while ΔNp63α-S385G cells (right panels) were transfected with an empty vector (triangles and squares) and ΔNp63α-FL cassette (WT-FL, diamonds). Cells were exposed to control medium (triangles) or 10μg/ml cisplatin (squares and diamonds) for indicated time periods. (A) Cell viability assay. Cell viability was assessed at 24, 48, 72, 96 and 120 h by MTT assay. Absorbance readings were taken using a SpectraMax M2e Microplate fluorescence reader (Molecular Devices) at 570- and 650- nm wavelengths. Experiments were performed in triplicate with SD as indicated (< 0.05). (B) Flow cytometry assay.
Discussion
Emerging high-throughput global protein interactions approaches (e.g., two-hybrid screens, protein arrays and mass spectrometry analysis) provide tools to identify numerous novel pathways/networks, leading to a better understanding of the systemic responses of cells to various stresses including new target chemo- and bio-therapies.Citation40-Citation44 Accumulating data suggests that there are multiple mechanisms that underlie the tumor cell response to cancer therapy and are implicated in chemoresistance.Citation41-Citation48 Among these molecular mechanisms, the regulation of RNA processing, stands out as an important mechanism that plays a key role in drug resistance.Citation49-Citation53 The Tp53 homolog ΔNp63α, as a member of the RNA transcription and splicing regulatory machinery, could contribute to cisplatin resistance via its interaction with the splicing regulatory factors.Citation13,Citation37
We previously found that wt-ΔNp63α cells exposed to cisplatin treatment displayed the ATM-dependent phosphorylation of ΔNp63α, rendering cells sensitive to cisplatin-induced cell death. At the same time, ΔNp63α-S385G cells failed to induce the cisplatin-mediated phosphorylation of ΔNp63α-S385G and render cells resistant to cisplatin exposure.Citation7,Citation54 Both p-ΔNp63α and non-p-ΔNp63α were shown to induce and downregulate a plethora of genes expressing mRNA and miRNA implicated in various pathways, ultimately leading to tumor cell death or cell survival.Citation7,Citation48,Citation54 As a continuation of our previous work, we further studied the role of protein interactions involving p-ΔNp63α and non-p-ΔNp63α proteins along with TP53-protein interactions in the SCC response to cisplatin resistance. Using a protein interaction chip approach, we first established the global Tp53 and TP63 interaction network comprising of 383 and 301 proteins, respectively. Using a combination of tandem affinity purification tagging and iTRAQ analysis, we further found that Tp53 and ΔNp63α interactomes include 444 and 310 proteins, respectively. We then explored the effect of cisplatin exposure on the differential protein-protein interactions of TP53, p-ΔNp63α and non-p-ΔNp63α in SCC cells. Altogether, our data shows that a number of proteins implicated in cell death/survival, DNA damage/signaling, chromatin remodeling/gene regulation, protein trafficking/degradation and RNA processing are potentially involved in SCC response to cisplatin treatment.
At first glance, we observed that a number of the specific p-ΔNp63α protein interactions were enriched in sensitive wt-ΔNp63α cells upon cisplatin exposure (e.g., SAP18, SIN3A, ATM, PRPF40B, DDIT3, ACIN1, etc.), while these interactions show the smallest iTRAQ ratios in the resistant ΔNp63α-S385G cells, suggesting the potential role for the ATM-dependent phosphorylation of ΔNp63α in such protein interactions (Tables S6 and S7). Although the cisplatin-induced Tp53 protein-protein interactions were enriched with proteins involved in cell death/apoptosis and cell cycle arrest pathways in SCC cells expressing wt-ΔNp63α or ΔNp63α-S385G, some of the specific TP53-protein interactions appeared to be potentially affected by p-ΔNp63α vs. non-p-ΔNp63α (e.g., STK11, KAT5, RPTOR, HMGB1, ASPP1, etc., Tables S4 and S5).
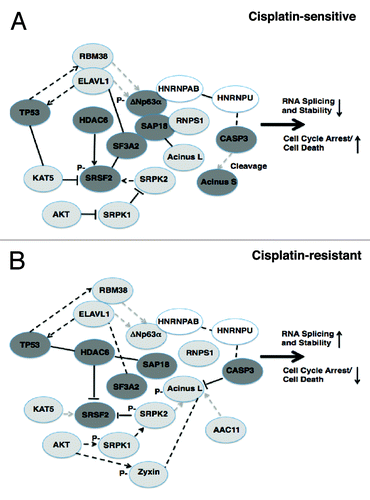
Because ΔNp63α functions as a regulator of RNA splicing through its interaction with the spliceosome components,Citation13,Citation37 we focused our follow-up studies on a number of proteins implicated in RNA processing/RNA stability and cell death, which stand out from the protein interaction chip array and iTRAQ data (e.g., SAP18, SF3A2, SRSF2, HDAC6, ACIN1 and RBM38) and collectively suggest that the p-ΔNp63α/non-p-ΔNp63α interplay play a role of the RNA splicing occurred in cisplatin-resistant SCC cells. depicts the potential role for p-ΔNp63α/non-p-ΔNp63α network in regulation of RNA processing/stability and, consequently, in control of cell death through the physical and functional interaction with the components of the nuclear speckle-localized splicing regulatory multiprotein ASAP complex and RBM38/ELAVL1 pathway.Citation33,Citation52,Citation55-Citation61 Our data suggests that the p-ΔNp63α protein is likely to induce an ASAP assembly followed by a potential reduction of RNA splicing and induction of cell death in sensitive wt-ΔNp63α cells upon cisplatin exposure, while non-p-ΔNp63α leads to an ASAP disassembly in resistant ΔNp63α-S385G cells, activation of RNA splicing and cell death reduction.
Figure 8. Schematic representation of the TP53 and TP63 interactions/activities leading to regulation of RNA splicing/stability and cell death in SCC cells upon cisplatin exposure. Protein physical and functional interaction networks were based on data obtained from STRING 9.0 protein-protein interaction and Pubmed literature databases. Solid lines represent protein interactions shown in this work, dashed lines derived from the database and literature. Solid arrows represent activation of the target level or function, while gray arrows represent repression of target level or function. Dark gray ovals represent targets enhancing cisplatin sensitivity, while light gray ovals represent targets enhancing cisplatin resistance. White ovals represent targets, which play a neutral role in cellular response to cisplatin.
Accumulating data have shown that splicing patterns can already be determined at the promoter of a gene, evidencing a coupling between transcription and alternative splicing.Citation30 In some human cancers, SRPK2 was recently found to bind and phosphorylate ACIN1 and redistribute it from the nuclear speckles to the nucleoplasm, resulting in cyclin A1 but not A2 upregulation, leading, in turn, to a regulation of cell cycle arrest.Citation31 Additionally, a modulation of CDKN1A pre-mRNA splicing was shown to affect the TP53-mediated apoptosis.Citation38 We found here that, in contrast to non-p-ΔNp63α, the cisplatin-induced p-ΔNp63α protein reduced cyclin A1 transcription and CDKN1A pre-mRNA splicing and cell viability, while induced ACIN1-mediated DNA fragmentation and cell cycle arrest.
Prior studies have shown that the ASAP-associated splicing factor SAP18 forms a protein complex with chromatin modifier SIN3 and histone deacetylase HDAC, thereby suggesting that members of the splicing regulatory apparatus might be also involved in the process of chromatin remodeling during apoptosis through a SIN3-HDAC-mediated deacetylation of histones.Citation62 We found here that p-ΔNp63α formed a complex with SAP18, leading to a subsequent association with SRSF2 and, ultimately, to a modulation of RNA splicing and induction of apoptosis in sensitive wt-ΔNp63α cells exposed to cisplatin (). Beatrice Eymin’s research team recently reported that in unstressed cells, histone acetyltransferase Tip60 (KAT5) acetylates SRSF2, thereby preventing SRPK2-mediated phosphorylation of SRSF2 and, in turn, induces a proteasome-dependent degradation of SRSF2 (reviewed in ref. Citation52 and Citation63–Citation65). However, in stressed cells, SRSF2 is deacetylated by HDAC6 and phosphorylated by SRPK2, subsequently increasing the SRSF2 protein level and leading to apoptosis through regulation of the splicing switch of CASP8 pre-mRNA.Citation52 We further found that non-p-ΔNp63α failed to bind SAP18, thereby releasing the latter from the ASAP complex and allowing it to bind HDAC6 and KAT5, leading to acetylation of SRSF2, activation of RNA splicing and modulation of cell cycle arrest or apoptosis ().
We then found that TP53/KAT5 protein interactions were enriched in sensitive wt-ΔNp63α cells (), while the association between Tp53 and KAT5 dramatically decreased in resistant ΔNp63α-S385G cells, while Tp53 formed complexes with HDAC6 and SAP18 (), potentially contributing to the cisplatin-induced cell cycle arrest and apoptosis or absence thereof, as evidenced by prior reports in references Citation63–Citation65.
Using both protein interaction array and iTRAQ technology, we found many Tp53 family member’s specific protein interactors associated with chromatin remodeling and epigenetic regulation (e.g., CHAF1A, KAT2B, CHD4, SIN3A and 3B, HDAC1, -2, -6, -7A and -9, MTA1 and -2, CTBP1, DNMT1, MECP2, NCOR2, etc.), RNA transcription (e.g., CITED2, TFDP1, SP1, E2F1, HIF1A, EP300, CREBBP, TRRAP, DDIT3, etc.) and RNA processing (RBM38, RNPS1, SAP18 and -30, SKP2, SRSF2, SF3A2, TRRAP, TACC1, etc.). We further defined that numerous proteins implicated in cancer development through regulation of cell cycle arrest, apoptosis and autophagy (AMBRA1, ATG14, BCL2, BCL2L1, BECN1, CDKN1B, CDK2, DAPK1, MTOR, RICTOR, RPTOR, RPS6KB1, UVRAG, etc.) may also participate in the TP53/TP63 protein interaction network, supporting the notion that multiple and sometimes opposing pathways regulated by the TP53/TP63 interactome might play a decisive role in the tumor cell response to chemotherapy.Citation66,Citation67 Therefore, future functional studies are needed to unravel the specific role of these protein interactions in the tumor cell response to cisplatin treatment, and to pinpoint novel protein targets for future small molecule-based and microRNA-based therapeutics beneficial for human cancer patients.Citation68-Citation72
Materials and Methods
Protein array chip analysis.
Custom protein binding microarray chips with 16,368 individual human GST-and His6-tagged full-length proteins (in duplicate) on the chip were produced by the Protein Microarray Core at the Johns Hopkins Medical Institutions, as described in reference Citation73. Recombinant GST-fused Tp53 (1–393 amino acid residues, H00007157-P02, Abnova) and TP63 (1–680 amino acid residues, H00008626-P01, Abnova) purified from the baculovirus-infected Sf9 insect cells were dissolved in 0.1 M sodium phosphate-0.2 M NaCl buffer, pH 7.5; 25 µg of protein was labeled with Alexa-Flour-647 microscale protein labeling kit (A30009, Molecular Probes/Invitrogen). Labeled proteins were purified by a BioGel P-30 Fine size exclusion column chromatography (BioRad) in phosphate-buffered saline (PBS). The extent of labeling (> 35–50%) was determined by measuring the absorbance at 280 nm and 647 nm. Before the incubation with the labeled bait proteins, the protein microarray chip slide was briefly washed with PBST (0.1% Tween-20) and blocked with PBST with 3% BSA for 60 min at 4°C in a circular shaker at 50 rpm. The slides were briefly rinsed with PBST, and ~400 ng of labeled protein in 100 µl PBST with 5 mM DTT (to minimize potential GST dimerization) was evenly applied on the slide. The slide was covered with Lifterslip and incubated in a humidity chamber for 90 min at 4°C. After three times 5 min washing with PBST, slide was briefly rinsed with distilled water and spun dry. After extensive washing (three times 5 min washing with PBST), the slides were spun dry, protected from light and scanned using a GenePix 4000B microarray scanner at the 635 nm. Generated images were analyzed with the GenePixTM Pro v6.0 (2007) image analysis software.
Fluorescence intensity measurements from each array element were compared with local background; background subtraction was performed; and correlation graphics were plotted to assess the reproducibility of the duplicate spots. Before normalization, spots showing defects were manually flagged, while “error” spots and spots with intensities lower than the sum of mean backgrounds were discarded. Signal distribution of the bait protein with a chip was plotted using Microsoft Excel software (v12.2.3, 2008 for Mac). Signals less than 10% of the maximal signal were considered as background ones and removed from the subsequent analysis. The signals in the peak of the curve were selected for bioinformatics analysis using GO (v1.1.974, 2010, www.geneontology.org) and IPA (v8.0, 2010) programs.
Cells, reagents and transfection.
SCC cell line (expressing wt-Tp53 and TP63) was isolated from primary tissue at the Department of Otolaryngology/Head and Neck Surgery of the Johns Hopkins University School of Medicine. Flp-In approach was used to generate the wt-ΔNp63α or ΔNp63α-S385G knock-in clones in the genomic DNA.Citation6,Citation37 Resulting SCC cells exclusively expressed the wt-ΔNp63α protein or ΔNp63α-S385G protein, which was confirmed by sequencing of the amplified cDNA clones from wt and mutated cells. These SCC cell clones were designated as wt-ΔNp63α cells and ΔNp63α-S385G cells. In some experiments, wt-ΔNp63α cells were transfected with the 100 ng ΔNp63α-S385G-FL expression cassette, while ΔNp63α-S385G cells were transfected with 100 ng ΔNp63α-FL expression cassette for 24 h using FuGENE6 (Roche). Cells were maintained in RPMI medium 1640 and 10% fetal bovine serum and incubated with control medium or 10 µg/ml cisplatin (Sigma) for the indicated time periods.
Antibodies, immunoprecipitation and immunoblotting.
We used mouse monoclonal antibodies to SF3A2 (H00008175-M01, Abnova) and RNPS1 (clone 7G8, H00010921-M05, Abnova), polyclonal antibodies to RBM38 (H00055544-B02, Abnova) and SAP18 (H00010284-D01, Abnova), rabbit polyclonal antibodies to HDAC6 (ab47181, Abcam), SRPK1 (ab90527, Abcam) and KAT5/Tip60 (ab59894, Abcam), a mouse monoclonal antibody to SRPK2 (ab67993, Abcam), a mouse monoclonal antibody to Acinus1 (clone 2005C3a, ab50827, Abcam), a mouse monoclonal antibody to SRSF2 (LS-C89240-100, LifeSpan Biosciences), a mouse monoclonal antibody to wt-Tp53 (clone BP53-12, #05-224, EMD/Millipore) and rabbit polyclonal antibody against β-actin (#AV40173, Sigma). A rabbit polyclonal antibody against ΔNp63 (anti-p40, PC373, residues 5–17 epitope) was purchased from EMD/Calbiochem. A custom rabbit polyclonal antibody against a phosphorylated peptide encompassing the ΔNp63α protein sequence (ATM motif, NKLPSV-pS-QLINPQQ, residues 379–392) was prepared and purified.Citation6 For immunoprecipitation, we used nuclear lysates obtained as follows: cells were resuspended in a hypotonic lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA) with protease inhibitors (Sigma); 0.5% Triton X-100 was then added and the nuclei were pelleted at 2,500 g for 10 min at 4°C. Nuclear pellets were then resuspended in the extract buffer (20 mM HEPES pH 7.9, 25% glycerol, 0.4 M NaCl, 0.1 mM EDTA, 0.1 mM EGTA), rocked for 15 min at 4°C and the supernatants were recovered by centrifugation at 10,000 g for 5 min at 4°C.
Quantitative (q)-PCR.
Total RNA was extracted from SCC cells with TRIzol reagent (Invitrogen). 1.2 µg of RNA was used as template for reverse transcription using the high-capacity cDNA Reverse Transcription kit (Applied Biosystems) as described (Huang et al. 2011). cDNA was dissolved in 20 µl of H2O. The cDNA levels of specific genes were amplified using the TaqMan® Gene expression assays [cyclin A1 (CCN1A), Hs00171105_m1; cyclin A2 (CCNA2), Hs00996788_m1 and control glyceraldehyde-3-phosphate-dehydrogenase (GAPDH), Hs02758991_g1] and TaqMan® Gene Expression Master Mix, 1-Pack (#4369016) obtained from Applied Biosystems. All qPCR reactions were performed in triplicate. For each experiment, the triplicate results of the qPCR were averaged, and this mean value was treated as a single statistical unit. The data were presented as means ± SD. Relative gene expression was normalized for the GAPDH housekeeping gene expression.
For splicing of intron 2 (between exons II and III) out of the CDKN1A pre-mRNA isoform 1, we used the following primers: sense, (+7,850) 5'-GCC TTG GCC TGC CCA AGC TCT ACC T-3' (+7,875) and antisense, (9,675) 5'-CAC TTG TCC GCT GGG TGG TAC CCT C-3' (+,9700), giving rise to the 1,850 bp PCR fragment for unspliced (US) product and 546 bp PCR fragment for spliced product.
Expression and purification of the NTAP protein complexes.
cDNA fragments for ΔNp63α, ΔNp63α-S385G and Tp53 were amplified using the following PCR primers: for ΔNp63α, sense, 5'-GGA TCC atg ttg tac ctg gaa aac a-3', antisense, 5'-caa aga gga ggg gga gtg aCA GCT G-3'; for Tp53, sense, 5'-GGA TCC atg gag gag ccg cag tca ga-3', antisense, 5'-aag ggc ctg act cag act gaC AGC TG-3'. Resulting PCR fragments were subcloned into the BamHI and XhoI sites of the pNTAP vector (Stratagene), thereby fusing the N-terminal regions of target proteins in frame to the SBP-tag located next to the CaBP tag. The pNTAP-ΔNp63α, pNTAP-ΔNp63α-S385G and pNTAP-Tp53 expression constructs were transiently introduced into SCC cells using Lipofectamine-2000 (Invitrogen) for 48 h, and the resulting cells were exposed to control medium or 10 µg/ml cisplatin. Cells grown on 20–30 150 mm dishes (to obtain 25–30 mg of protein per sample) were lysed with buffer A (50 mM Tris, pH 7.5, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5% Brij-50, 1 mM PMSF, 0.5 mM NaF, 0.1 mM Na3VO4, 2x complete protease inhibitor cocktail), sonicated and clarified for 30 min at 15,000 g. Protein complexes were purified from the supernatants using NTAP system (#240103, Stratagene/Agilent Technology) under native conditions.Citation25,Citation27
iTRAQ labeling and liquid chromatography-double mass spectrometry (LC-MS/MS).
Protein complexes were precipitated with trichloroacetic acid (TCA) and desalted. The trypsin-digested peptides, were labeled with the isobaric tags 113 and 114 (for samples obtained from cells exposed to control media) and 115–118 (for samples obtained from cells exposed to cisplatin) using the iTRAQ Reagents Application Kit (#4374321, Applied Biosystems). Samples were then mixed, dried and fractionated by strong cationic exchange (SCX) chromatography on an Agilent HPLC system. Each SCX fraction was separated on a C18 column using 5–40% (90% acetonitrile in 0.1% formic acid) gradient over 60 min. Eluted peptides were sprayed directly into an LTQ Orbitrap Velos mass-spectrometer (Thermo-Scientific).
Data analysis.
The MS/MS spectra were extracted and searched against the RefSeq 40 database using Mascot (Matrix Science, version 10/11/2009) through Proteome Discoverer software (v1.1, Thermo-Scientific). The iTRAQ ratios were normalized by total protein, and only proteins identified with ratios > 1.2 or < 0.8 were considered as potential differential interactors. iTRAQ ratios were normalized to the median ratio using the following formula: iTRAQ ratio 1/4 ratio/median iTRAQ ratio of all found pairs. Both correction and normalization were performed using GPS Explorer software v3.6. All protein quantifications were based on the spectral counts observed for each protein. Spectral counts for each protein were averaged and used to calculate fold enrichment over the control. All tables and graphs were generated using Microsoft Excel.
Protein interaction data sets were analyzed using IPA software, v8.0 (2010, www.ingenuity.com) and STRING v9.0 protein interaction database (2009, www.string.embl.de). Analysis of entire protein data sets using IPA Knowledge Base identified the most significant biological functions/diseases, canonical pathways and networks. Significances for functional enrichment of specific genes and the ranking scores for each network were computed by IPA based on a right-tailed Fisher’s exact test showing (as the negative log of the probability) that the number of genes in the network is not due to random chance using all input genes as a reference set. IPA-derived functions and pathways were selected as those with a probability higher than a threshold (p < 0.05).
DNA fragmentation assay.
Cell pellets were lysed on ice with a 10 mM TRIS-HCl (pH 8.0), 10 mM EDTA and 0.5% Triton X-100 for 15 min. The lysate was centrifuged at 12,000x g for 15 min to separate soluble (fragmented) from pellet (intact genomic) DNA. Soluble DNA was treated with RNA-ase A (50 µg/ml) at 37°C for 1 h, followed by treatment with proteinase K (100 µg/ml) in 0.5% SDS at 50°C for 2 h. The residual material was extracted with phenol/chloroform, precipitated in ethanol, resolved by a 1.8% agarose gel electrophoresis, stained with ethidium bromide and the gels were photographed under UV light.
Flow cytometry cell cycle analysis.
Cells were harvested, washed in PBS and fixed in methanol (4): acetic acid (1) solution for 60 min at 4°C. Cells were then incubated in 500 µl of staining solution (50 µg/µl of propidium iodide, 50 µg/µl of RNA-ase, 0.1% Triton X-100 in 1 PBS) for 1 h at 4°C and analyzed by flow cytometry performed on the FACSCalibur instrument (BD Biosciences).
Cell viability assay.
Cell viability was analyzed by measuring the capacity of cells to reduce 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT, American Tissue Culture Collection) to formazan. Cells were plated at 20–30% confluence per well of a 6-well plate, and the cell viability was assayed at 24 h, 48 h, 72 h, 96 h and 120 h by MTT cell viability assay in triplicate by monitoring the absorbance at 570 and 650 nm wavelengths using a SpectraMax M2e Microplate fluorescence reader (Molecular Devices), as previously described in references Citation48 and Citation54.
Statistical analysis.
The data represent mean ± SD from three independent experiments and the statistical analysis was performed by t-test at a significance level of p < 0.05.
Concluding Remarks
Our observations suggest that Tp53 and ΔNp63α are involved in numerous protein-protein interactions, which are likely to be implicated in response by tumor cells to cisplatin exposure. Phosphorylated (p)-ΔNp63α binds to the splicing complex leading to repression of mRNA splicing and activation of ACIN1-mediated cell death pathway. In contrast to p-ΔNp63α, non-p-ΔNp63α fails to bind the critical members of the splicing complex, thereby leading to activation of RNA splicing and reduction of cell death pathway. Overall, our studies provide an integrated proteomic platform in making a case for the role of the p53/p63 interactome in cisplatin chemoresistance.
| Abbreviations: | ||
| ATM | = | ataxia telangiectasia mutated |
| CIS | = | cisplatin |
| Con | = | control, DTT, dithiothreitol, GO, Gene Ontology |
| GST | = | glutathione-S-transferase |
| iTRAQ | = | isobaric tags for relative and absolute quantification |
| IPA | = | Ingenuity Pathways Analysis |
| LC-MS/MS | = | liquid chromatography-double mass spectrometry |
| NTAP | = | N-terminal mammalian tandem affinity purification |
| p | = | phosphorylated |
| quantitative PCR | = | qPCR |
| SCC | = | squamous cell carcinoma |
| SCX | = | strong cationic exchange |
| SP | = | spliced |
| TP | = | tumor protein |
| US | = | unspliced |
Additional material
Download Zip (2.3 MB)Acknowledgments
This work was supported in part by the Flight Attendant Research Institutions grant (#082469 to E.A.R.), and by National Cancer Institute grants K01-CA164092 and U01-CA84986 (R.G-P.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note
Supplemental materials can be found at: www.landesbioscience.com/journals/cc/article/20863
Related Research Data
References
- Helmbach H, Kern MA, Rossmann E, Renz K, Kissel C, Gschwendt B, et al. Drug resistance towards etoposide and cisplatin in human melanoma cells is associated with drug-dependent apoptosis deficiency. J Invest Dermatol 2002; 118:923 - 32; http://dx.doi.org/10.1046/j.1523-1747.2002.01786.x; PMID: 12060385
- Zangen R, Ratovitski EA, Sidransky D. DeltaNp63α levels correlate with clinical tumor response to cisplatin. Cell Cycle 2005; 4:1313 - 5; http://dx.doi.org/10.4161/cc.4.10.2066; PMID: 16123597
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7:573 - 84; http://dx.doi.org/10.1038/nrc2167; PMID: 17625587
- Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. P63 and p73 are required for TP53-dependent apoptosis in response to DNA damage. Nature 2002; 416:560 - 4; http://dx.doi.org/10.1038/416560a; PMID: 11932750
- Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006; 9:45 - 56; http://dx.doi.org/10.1016/j.ccr.2005.12.013; PMID: 16413471
- Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski E, et al. ATM kinase is a master switch for the Δ Np63 α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008; 7:2846 - 55; http://dx.doi.org/10.4161/cc.7.18.6627; PMID: 18769144
- Huang Y, Chuang AY, Romano RA, Liegeois NJ, Sinha S, Trink B, et al. Phospho-ΔNp63α/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle 2010; 9:332 - 42; http://dx.doi.org/10.4161/cc.9.2.10432
- Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ 2010; 17:901 - 11; http://dx.doi.org/10.1038/cdd.2010.35; PMID: 20379196
- Tozluoğlu M, Karaca E, Haliloglu T, Nussinov R. Cataloging and organizing p73 interactions in cell cycle arrest and apoptosis. Nucleic Acids Res 2008; 36:5033 - 49; http://dx.doi.org/10.1093/nar/gkn481; PMID: 18660513
- Ratovitski EA, Patturajan M, Hibi K, Trink B, Yamaguchi K, Sidransky D. p53 associates with and targets Δ Np63 into a protein degradation pathway. Proc Natl Acad Sci USA 2001; 98:1817 - 22; http://dx.doi.org/10.1073/pnas.98.4.1817; PMID: 11172034
- Zeng SX, Dai MS, Keller DM, Lu H. SSRP1 functions as a co-activator of the transcriptional activator p63. EMBO J 2002; 21:5487 - 97; http://dx.doi.org/10.1093/emboj/cdf540; PMID: 12374749
- Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, et al. DeltaNp63 induces β-catenin nuclear accumulation and signaling. Cancer Cell 2002; 1:369 - 79; http://dx.doi.org/10.1016/S1535-6108(02)00057-0; PMID: 12086851
- Fomenkov A, Huang YP, Topaloglu O, Brechman A, Osada M, Fomenkova T, et al. P63 α mutations lead to aberrant splicing of keratinocyte growth factor receptor in the Hay-Wells syndrome. J Biol Chem 2003; 278:23906 - 14; http://dx.doi.org/10.1074/jbc.M300746200; PMID: 12692135
- Huang YP, Wu G, Guo Z, Osada M, Fomenkov T, Park HL, et al. Altered sumoylation of p63α contributes to the split-hand/foot malformation phenotype. Cell Cycle 2004; 3:1587 - 96; http://dx.doi.org/10.4161/cc.3.12.1290; PMID: 15539951
- MacPartlin M, Zeng S, Lee H, Stauffer D, Jin Y, Thayer M, et al. p300 regulates p63 transcriptional activity. J Biol Chem 2005; 280:30604 - 10; http://dx.doi.org/10.1074/jbc.M503352200; PMID: 15965232
- Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci USA 2006; 103:12753 - 8; http://dx.doi.org/10.1073/pnas.0603449103; PMID: 16908849
- Wang N, Guo L, Rueda BR, Tilly JL. Cables1 protects p63 from proteasomal degradation to ensure deletion of cells after genotoxic stress. EMBO Rep 2010; 11:633 - 9; http://dx.doi.org/10.1038/embor.2010.82; PMID: 20559324
- Chatterjee A, Chang X, Sen T, Ravi R, Bedi A, Sidransky D. Regulation of p53 family member isoform DeltaNp63alpha by the nuclear factor-kappaB targeting kinase IkappaB kinase beta. Cancer Res 2010; 70:1419 - 29; http://dx.doi.org/10.1158/0008-5472.CAN-09-2613; PMID: 20145131
- Amoresano A, Di Costanzo A, Leo G, Di Cunto F, La Mantia G, Guerrini L, et al. Identification of DeltaNp63α protein interactions by mass spectrometry. J Proteome Res 2010; 9:2042 - 8; http://dx.doi.org/10.1021/pr9011156; PMID: 20085233
- Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, et al. Global analysis of protein activities using proteome chips. Science 2001; 293:2101 - 5; http://dx.doi.org/10.1126/science.1062191; PMID: 11474067
- Bai Y, Markham K, Chen F, Weerasekera R, Watts J, Horne P, et al. The in vivo brain interactome of the amyloid precursor protein. Mol Cell Proteomics 2008; 7:15 - 34; http://dx.doi.org/10.1074/mcp.M700077-MCP200; PMID: 17934213
- Schweitzer B, Predki P, Snyder M. Microarrays to characterize protein interactions on a whole-proteome scale. Proteomics 2003; 3:2190 - 9; http://dx.doi.org/10.1002/pmic.200300610; PMID: 14595818
- Schnack C, Hengerer B, Gillardon F. Identification of novel substrates for Cdk5 and new targets for Cdk5 inhibitors using high-density protein microarrays. Proteomics 2008; 8:1980 - 6; http://dx.doi.org/10.1002/pmic.200701063; PMID: 18491313
- O’Connell DJ, Bauer MC, O’Brien J, Johnson WM, Divizio CA, O’Kane SL, et al. Integrated protein array screening and high throughput validation of 70 novel neural calmodulin-binding proteins. Mol Cell Proteomics 2010; 9:1118 - 32; http://dx.doi.org/10.1074/mcp.M900324-MCP200; PMID: 20068228
- McFarland MA, Ellis CE, Markey SP, Nussbaum RL. Proteomics analysis identifies phosphorylation-dependent α-synuclein protein interactions. Mol Cell Proteomics 2008; 7:2123 - 37; http://dx.doi.org/10.1074/mcp.M800116-MCP200; PMID: 18614564
- Pflieger D, Jünger MA, Müller M, Rinner O, Lee H, Gehrig PM, et al. Quantitative proteomic analysis of protein complexes: concurrent identification of interactors and their state of phosphorylation. Mol Cell Proteomics 2008; 7:326 - 46; http://dx.doi.org/10.1074/mcp.M700282-MCP200; PMID: 17956857
- Bousquet-Dubouch MP, Baudelet E, Guérin F, Matondo M, Uttenweiler-Joseph S, Burlet-Schiltz O, et al. Affinity purification strategy to capture human endogenous proteasome complexes diversity and to identify proteasome-interacting proteins. Mol Cell Proteomics 2009; 8:1150 - 64; http://dx.doi.org/10.1074/mcp.M800193-MCP200; PMID: 19193609
- Chen LC, Chung IC, Hsueh C, Tsang NM, Chi LM, Liang Y, et al. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ 2010; 17:1463 - 73; http://dx.doi.org/10.1038/cdd.2010.24; PMID: 20224598
- Kaake RM, Wang X, Huang L. Profiling of protein interaction networks of protein complexes using affinity purification and quantitative mass spectrometry. Mol Cell Proteomics 2010; 9:1650 - 65; http://dx.doi.org/10.1074/mcp.R110.000265; PMID: 20445003
- Moore MJ, Wang Q, Kennedy CJ, Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010; 142:625 - 36; http://dx.doi.org/10.1016/j.cell.2010.07.019; PMID: 20705336
- Jang SW, Yang SJ, Ehlén A, Dong S, Khoury H, Chen J, et al. Serine/arginine protein-specific kinase 2 promotes leukemia cell proliferation by phosphorylating acinus and regulating cyclin A1. Cancer Res 2008; 68:4559 - 70; http://dx.doi.org/10.1158/0008-5472.CAN-08-0021; PMID: 18559500
- Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev 2009; 23:482 - 95; http://dx.doi.org/10.1101/gad.1752109; PMID: 19240134
- Schwerk C, Prasad J, Degenhardt K, Erdjument-Bromage H, White E, Tempst P, et al. ASAP, a novel protein complex involved in RNA processing and apoptosis. Mol Cell Biol 2003; 23:2981 - 90; http://dx.doi.org/10.1128/MCB.23.8.2981-2990.2003; PMID: 12665594
- Joselin AP, Schulze-Osthoff K, Schwerk C. Loss of Acinus inhibits oligonucleosomal DNA fragmentation but not chromatin condensation during apoptosis. J Biol Chem 2006; 281:12475 - 84; http://dx.doi.org/10.1074/jbc.M509859200; PMID: 16537548
- Jiang ZH, Zhang WJ, Rao Y, Wu JY. Regulation of Ich-1 pre-mRNA alternative splicing and apoptosis by mammalian splicing factors. Proc Natl Acad Sci USA 1998; 95:9155 - 60; http://dx.doi.org/10.1073/pnas.95.16.9155; PMID: 9689050
- Fushimi K, Ray P, Kar A, Wang L, Sutherland LC, Wu JY. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl Acad Sci USA 2008; 105:15708 - 13; http://dx.doi.org/10.1073/pnas.0805569105; PMID: 18840686
- Huang YP, Kim Y, Li Z, Fomenkov T, Fomenkov A, Ratovitski EA. AEC-associated p63 mutations lead to alternative splicing/protein stabilization of p63 and modulation of Notch signaling. Cell Cycle 2005; 4:1440 - 7; http://dx.doi.org/10.4161/cc.4.10.2086; PMID: 16177572
- Chen Y, Zhang L, Jones KA. SKIP counteracts p53-mediated apoptosis via selective regulation of p21Cip1 mRNA splicing. Genes Dev 2011; 25:701 - 16; http://dx.doi.org/10.1101/gad.2002611; PMID: 21460037
- Edmond V, Brambilla C, Brambilla E, Gazzeri S, Eymin B. SRSF2 is required for sodium butyrate-mediated p21(WAF1) induction and premature senescence in human lung carcinoma cell lines. Cell Cycle 2011; 10:1968 - 77; http://dx.doi.org/10.4161/cc.10.12.15825; PMID: 21555914
- Albers M, Kranz H, Kober I, Kaiser C, Klink M, Suckow J, et al. Automated yeast two-hybrid screening for nuclear receptor-interacting proteins. Mol Cell Proteomics 2005; 4:205 - 13; http://dx.doi.org/10.1074/mcp.M400169-MCP200; PMID: 15604093
- Gingras AC, Caballero M, Zarske M, Sanchez A, Hazbun TR, Fields S, et al. A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Mol Cell Proteomics 2005; 4:1725 - 40; http://dx.doi.org/10.1074/mcp.M500231-MCP200; PMID: 16085932
- Stewart JJ, White JT, Yan X, Collins S, Drescher CW, Urban ND, et al. Proteins associated with Cisplatin resistance in ovarian cancer cells identified by quantitative proteomic technology and integrated with mRNA expression levels. Mol Cell Proteomics 2006; 5:433 - 43; http://dx.doi.org/10.1074/mcp.M500140-MCP200; PMID: 16319398
- Tu LC, Yan X, Hood L, Lin B. Proteomics analysis of the interactome of N-myc downstream regulated gene 1 and its interactions with the androgen response program in prostate cancer cells. Mol Cell Proteomics 2007; 6:575 - 88; http://dx.doi.org/10.1074/mcp.M600249-MCP200; PMID: 17220478
- Chavez JD, Hoopmann MR, Weisbrod CR, Takara K, Bruce JE. Quantitative proteomic and interaction network analysis of cisplatin resistance in HeLa cells. PLoS ONE 2011; 6:e19892; http://dx.doi.org/10.1371/journal.pone.0019892; PMID: 21637840
- Gatti L, Zunino F. Overview of tumor cell chemoresistance mechanisms. Methods Mol Med 2005; 111:127 - 48; PMID: 15911977
- Righetti PG, Castagna A, Antonioli P, Cecconi D, Campostrini N, Righetti SC. Proteomic approaches for studying chemoresistance in cancer. Expert Rev Proteomics 2005; 2:215 - 28; http://dx.doi.org/10.1586/14789450.2.2.215; PMID: 15892566
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31:1869 - 83; http://dx.doi.org/10.1038/onc.2011.384; PMID: 21892204
- Sen T, Sen N, Brait M, Begum S, Chatterjee A, Hoque MO, et al. ΔNp63α confers tumor cell resistance to cisplatin treatment through the AKT transcriptional regulation. Cancer Res 2011; 71:1167 - 76; http://dx.doi.org/10.1158/0008-5472.CAN-10-1481; PMID: 21266360
- Chandler DS, Singh RK, Caldwell LC, Bitler JL, Lozano G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res 2006; 66:9502 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-4271; PMID: 17018606
- Hayes GM, Carrigan PE, Beck AM, Miller LJ. Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res 2006; 66:3819 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-05-4065; PMID: 16585209
- Ezponda T, Pajares MJ, Agorreta J, Echeveste JI, López-Picazo JM, Torre W, et al. The oncoprotein SF2/ASF promotes non-small cell lung cancer survival by enhancing survivin expression. Clin Cancer Res 2010; 16:4113 - 25; http://dx.doi.org/10.1158/1078-0432.CCR-10-0076; PMID: 20682707
- Edmond V, Moysan E, Khochbin S, Matthias P, Brambilla C, Brambilla E, et al. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J 2011; 30:510 - 23; http://dx.doi.org/10.1038/emboj.2010.333; PMID: 21157427
- Shkreta L, Michelle L, Toutant J, Tremblay ML, Chabot B. The DNA damage response pathway regulates the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem 2011; 286:331 - 40; http://dx.doi.org/10.1074/jbc.M110.162644; PMID: 20980256
- Huang YP, Chuang A, Hao H, Talbot CC, Trink B, Sidransky D, et al. Phospho-ΔNp63α is a key regulator of cisplatin-induced microRNAome in head and neck squamous cell carcinoma cells. Cell Death Differ 2011; 18:1220 - 30; http://dx.doi.org/10.1038/cdd.2010.188; PMID: 21274007
- Blaustein M, Pelisch F, Tanos T, Muñoz MJ, Wengier D, Quadrana L, et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol 2005; 12:1037 - 44; http://dx.doi.org/10.1038/nsmb1020; PMID: 16299516
- Hu Y, Yao J, Liu Z, Liu X, Fu H, Ye K. Akt phosphorylates acinus and inhibits its proteolytic cleavage, preventing chromatin condensation. EMBO J 2005; 24:3543 - 54; http://dx.doi.org/10.1038/sj.emboj.7600823; PMID: 16177823
- Singh KK, Erkelenz S, Rattay S, Dehof AK, Hildebrandt A, Schulze-Osthoff K, et al. Human SAP18 mediates assembly of a splicing regulatory multiprotein complex via its ubiquitin-like fold. RNA 2010; 16:2442 - 54; http://dx.doi.org/10.1261/rna.2304410; PMID: 20966198
- Mazan-Mamczarz K, Galbán S, López de Silanes I, Martindale JL, Atasoy U, Keene JD, et al. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA 2003; 100:8354 - 9; http://dx.doi.org/10.1073/pnas.1432104100; PMID: 12821781
- Shu L, Yan W, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Genes Dev 2006; 20:2961 - 72; http://dx.doi.org/10.1101/gad.1463306; PMID: 17050675
- Cho SJ, Zhang J, Chen X. RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Res 2010; 38:2256 - 67; http://dx.doi.org/10.1093/nar/gkp1229; PMID: 20064878
- Zhang J, Jun Cho S, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci USA 2010; 107:9614 - 9; http://dx.doi.org/10.1073/pnas.0912594107; PMID: 20457941
- Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P, Reinberg D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1). Mol Cell Biol 2002; 22:835 - 48; http://dx.doi.org/10.1128/MCB.22.3.835-848.2002; PMID: 11784859
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006; 24:841 - 51; http://dx.doi.org/10.1016/j.molcel.2006.11.026; PMID: 17189187
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006; 24:827 - 39; http://dx.doi.org/10.1016/j.molcel.2006.11.021; PMID: 17189186
- Miyamoto N, Izumi H, Noguchi T, Nakajima Y, Ohmiya Y, Shiota M, et al. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. J Biol Chem 2008; 283:18218 - 26; http://dx.doi.org/10.1074/jbc.M802332200; PMID: 18458078
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22:181 - 5; http://dx.doi.org/10.1016/j.ceb.2009.12.001; PMID: 20044243
- Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Mariño G, Galluzzi L, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011; 10:2763 - 9; http://dx.doi.org/10.4161/cc.10.16.16868; PMID: 21775823
- Sen T, Chang X, Sidransky D, Chatterjee A. Regulation of ΔNp63α by NFκΒ. Cell Cycle 2010; 9:4841 - 7; http://dx.doi.org/10.4161/cc.9.24.14093; PMID: 21088498
- Bagnoli M, De Cecco L, Granata A, Nicoletti R, Marchesi E, Alberti P, et al. Identification of a chrXq27.3 microRNA cluster associated with early relapse in advanced stage ovarian cancer patients. Oncotarget 2011; 2:1265 - 78; PMID: 22246208
- Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, et al. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget 2011; 2:1203 - 17; PMID: 22203497
- Ory B, Ellisen LW. A microRNA-dependent circuit controlling p63/p73 homeostasis: p53 family cross-talk meets therapeutic opportunity. Oncotarget 2011; 2:259 - 64; PMID: 21436470
- Nijwening JH, Kuiken HJ, Beijersbergen RL. Screening for modulators of cisplatin sensitivity: unbiased screens reveal common themes. Cell Cycle 2011; 10:380 - 6; http://dx.doi.org/10.4161/cc.10.3.14642; PMID: 21239890
- Jeong JS, Jiang L, Albino E, Marrero J, Rho HS, Hu J, et al. Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol Cell Proteomics 2012; In press http://dx.doi.org/10.1074/mcp.O111.016253; PMID: 22307071