Abstract
The limitations of revolutionary new mutation-specific inhibitors of BRAFV600E include the universal recurrence seen in melanoma patients treated with this novel class of drugs. Recently, our lab showed that simultaneous activation of the Wnt/β-catenin signaling pathway and targeted inhibition of BRAFV600E by PLX4720 synergistically induces apoptosis across a spectrum of BRAFV600E melanoma cell lines. As a follow-up to that study, treatment of BRAF-mutant and NRAS-mutant melanoma lines with WNT3A and the MEK inhibitor AZD6244 also induces apoptosis. The susceptibility of BRAF-mutant lines and NRAS-mutant lines to apoptosis correlates with negative regulation of Wnt/β-catenin signaling by ERK/MAPK signaling and dynamic decreases in abundance of the downstream scaffolding protein, AXIN1. Apoptosis-resistant NRAS-mutant lines can sensitize to AZD6244 by pretreatment with AXIN1 siRNA, similar to what we previously reported in BRAF-mutant cell lines. Taken together, these findings indicate that NRAS-mutant melanoma share with BRAF-mutant melanoma the potential to regulate apoptosis upon MEK inhibition through WNT3A and dynamic regulation of cellular AXIN1. Understanding the cellular context that makes melanoma cells susceptible to this combination treatment will contribute to the study and development of novel therapeutic combinations that may lead to more durable responses.
Introduction
Melanoma represents a devastating disease responsible for 80% of deaths resulting from skin cancer.Citation1 The 5-y survival rate of 15% illustrates the poor prognosis of for melanoma patients with metastatic disease.Citation2 Making matters worse, melanoma incidence continues to rise at a rate faster than any other cancer.Citation3 Our growing knowledge of the molecular drivers of melanoma provides perhaps the best hope for identifying novel therapeutics to this disease. Recently, the first mutation-targeted chemotherapeutic agent for any cancer, vemurafenib (trade name Zelboraf), has exhibited promising results in the treatment of patients with metastatic melanoma. Vemurafenib (previously called PLX4032) specifically inhibits a constitutively active, mutant form of BRAF,Citation4-Citation7 an oncogene that helps drive approximately 50% of melanoma.Citation8,Citation9 While this revolutionary targeted approach to cancer therapy resulted in tumor reduction in almost all treated patients, recurrence was inevitable, with half of patients relapsing within 6 mo. Patients treated with vemurafenib exhibited a median duration of progression-free survival of 7 mo and an increase in 2-y overall survival to 23.5%.Citation10-Citation12 While these results are extremely promising compared with previous melanoma therapies, the universal development of resistance indicates that monotherapy is unlikely to produce clinically durable outcomes. The potent effect of targeted BRAF inhibitors on melanoma cells provides a context where additional therapeutic strategies applied in parallel or in series could lead to durable clinical outcomes.
Preliminary evidence from melanocyte development and patient-derived human models implicate the Wnt/β-catenin signaling pathway in melanoma pathogenesis. Wnts are secreted glycoproteins that can transduce their signal into a cell in a variety of ways. The canonical class of Wnt/β-catenin ligands, including WNT1 and WNT3A, can bind Frizzled and LRP5/6 co-receptors.Citation13 This ligand-mediated binding action inhibits the degradation of cytoplasmic β-catenin (encoded by CTNNB1), which subsequently accumulates, translocates to the nucleus and directs the expression of a diverse array of genes in a cell type-specific manner. During melanocyte development, Wnt/β-catenin signaling is necessary and sufficient to drive neural crest cells toward a melanocyte cell fate, in large part through direct regulation of transcriptional targets, such as microphthalmia transcription factor (encoded by MITF).Citation14-Citation18 During melanoma pathogenesis, the observed presence of nuclear β-catenin in the majority of benign nevi, along with the loss of nuclear β-catenin seen with melanoma progression,Citation19-Citation21 support the hypothesis that maintenance of homeostatic levels of Wnt/β catenin signaling is important in this particular context. Furthermore, high cytoplasmic and/or nuclear β-catenin staining of patient tumor biopsies correlates with increased patient survival, decreased metastasis recurrence and decreased expression of proliferation markers.Citation19,Citation22,Citation23 Supportingly, high Wnt/β-catenin signaling mediated by overexpression of WNT3A inhibits tumor growth in mouse models of melanoma, accompanied by an increased expression of melanocyte-associated gene targets.Citation22,Citation24
In light of the inhibitory role for Wnt/β-catenin signaling in this context as well as existing clinical efforts to target BRAF-mediated signaling in melanoma, we examined the combinatorial effects of Wnt/β-catenin activation with ERK/MAPK inhibition. In doing so, we found that simultaneous activation of Wnt/β-catenin signaling pathway and inhibition of BRAFV600E by PLX4720 synergistically induced apoptosis in a selection of melanoma cell lines. Furthermore, overexpression of WNT3A in human A375 cells in combination with PLX4720 treatment resulted in significant inhibition of tumor growth in relevant mouse models of melanoma.Citation24 Not only did Wnt/β-catenin signaling enhance PLX4720-mediated apoptosis, it is also unexpectedly required for this apoptosis, which was inhibited in cell lines sensitive to PLX4720 alone by pretreatment with siRNA targeting β-catenin.Citation24
Although a subset of cell lines exhibited significant apoptosis with the combination of WNT3A and PLX4720, several cell lines showed no response to this combination treatment.Citation24 Further analysis revealed that apoptotic sensitivity correlated with dynamic loss of AXIN1, a negative regulator of Wnt/β-catenin signaling.Citation24 In support of the central role of AXIN1 in mediating this response, the cell lines that did not exhibit significant apoptosis with WNT3A and PLX4720 could be sensitized to this combination by siRNA-mediated knockdown of AXIN1, thus conferring a significant apoptotic response. Taken together, these findings define a preliminary context wherein Wnt/β-catenin signaling and the loss of cellular AXIN1 abundance can promote BRAF-inhibitor-mediated apoptosis.
Shared Characteristics of Susceptible and Resistant NRAS- and BRAF-Mutant Lines
Our initial work was performed in BRAF-mutant melanoma lines, as approximately 45% of melanomas carry BRAF-activating mutations, which activate the ERK/MAPK pathway.Citation9 However, an additional 20% of melanoma harbor activating NRAS mutations, which likewise, activate ERK/MAPK signaling.Citation9 Based on our initial finding that Wnt/β-catenin signaling cooperates with targeted BRAFV600E inhibitors to promote apoptosis in BRAF-mutant melanoma, we hypothesized that WNT3A treatment would also cooperate with ERK/MAPK inhibitors in a subset of NRAS-mutant melanomas. As predicted, melanoma cell lines harboring either the BRAF or the NRAS mutation exhibit variations in sensitivity to the combination of WNT3A and a small molecule MEK inhibitor, with several lines of each genetic class exhibiting relative resistance. As with BRAF-mutant melanoma, these data show that some NRAS-mutant melanomas, are inherently susceptible to apoptosis with the combination of WNT3A and MEK inhibition (), while some exhibit no significant response.
Figure 1. Regulation of Wnt/β-catenin signaling by ERK/MAPK and loss of AXIN1 protein predict apoptotic response to combined Wnt/β-catenin activation and MEK inhibition. (A) Flow cytometry detection of cleaved PARP was used to measure apoptosis in a panel of cell lines harboring activating BRAF or NRAS mutations as indicated. (B) Enhancement of Wnt/β-catenin signaling by MEK inhibition was measured by quantitative PCR measurements of the endogenous Wnt/β-catenin target gene AXIN2. Data are expressed as copies of AXIN2 per 106 copies of ACTB. (C) Immunoblot analysis of same cell lines for loss of AXIN1 protein and for loss of phospho-ERK (ppERK) upon inhibition of MEK by AZD6244. In (A), (B) and (C) cells were treated with the indicated conditions for 24 h. Two uM AZD6244 was used where indicated. For (A) and (B), columns and error bars represent the mean and SEM, respectively, of three biologic replicates. *p < 0.05 by one-way ANOVA with Tukey’s post-test. (C) is a representative immunoblot of three biologic replicates.
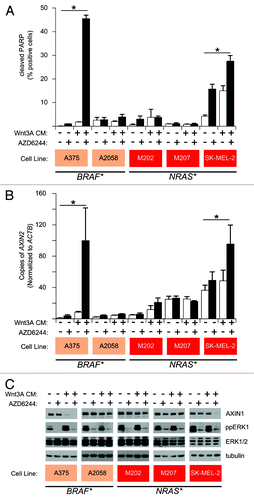
In apoptosis-sensitive lines (A375 and SK-MEL-2), ERK/MAPK pathway inhibition increased WNT3A-dependent transcription, while in apoptosis-resistant lines (A2058, M202 and M207) ERK/MAPK inhibition had no effect (). As seen in BRAFV600E melanoma cell lines, AXIN1 abundance also correlates with susceptibility to apoptosis in NRAS-mutant lines (). Finally, apoptosis-resistant NRAS-mutant melanoma cell lines can be sensitized to apoptosis by pretreatment with AXIN1 siRNA (). These findings point to AXIN1 expression as the central regulator of apoptosis in both NRAS- and BRAF-mutant melanoma cell lines. ERK/MAPK signaling regulates AXIN1 steady-state abundance and Wnt/β-catenin signal transduction in apoptosis-sensitive lines, implicating a common mechanism of sensitivity across multiple mutational backgrounds (). The observation that AXIN1 siRNA enhances apoptosis following MEK inhibitor treatment in both mutant backgrounds suggests that apoptosis-resistant lines can be made sensitive, regardless of differences in NRAS- or BRAF-mutation ().
Figure 2. AXIN1 depletion sensitizes NRAS- and BRAF-mutant melanoma cells to apoptosis mediated by MEK inhibition. Knockdown of AXIN1 by siRNA sensitizes A2058, M202 and M207 cells to AZD6244 induced apoptosis. Immunoblots show lysates from the indicated cell lines transfected with either control or siRNA targeting AXIN1 and treated with the indicated conditions for 24 h. Apoptosis, loss of AXIN1, and inhibition of MEK by AZD6244 were monitored by immunoblots for cleaved PARP, AXIN1 and ppERK, respectively.
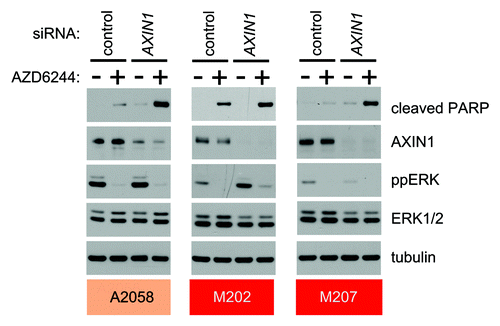
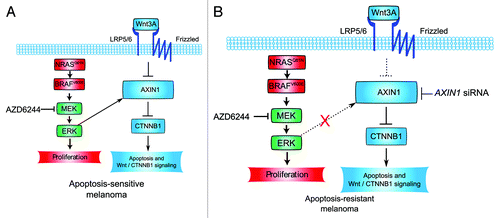
Figure 3. Melanoma sensitivity and resistance to WNT3A- and AZD6244-mediated apoptosis: a working model. (A) NRAS- and BRAF-mutant melanoma can both undergo apoptosis in response to WNT3A and AZD6244 treatment. In sensitive lines, WNT3A-treatment and MEK-inhibition cooperate to reduce AXIN1 protein levels. Downstream of AXIN1 loss, apoptosis occurs, and Wnt/β-catenin target gene transcription is synergistically activated. β-catenin is required for both apoptosis and target gene transcription. How exactly ERK/MAPK signaling regulates AXIN1 levels remains unresolved. (B) NRAS- and BRAF-mutant melanoma can both resist apoptosis in response to WNT3A and AZD6244 treatment. In this context, MEK inhibition by AZD6244 does not cooperate with WNT3A treatment to inhibit AXIN1 expression. Additionally, MEK inhibition does not change the cells responsiveness to WNT3A. AXIN1 siRNA is sufficient to potentiate AZD6244-mediated apoptosis. It remains unknown what factor, or set of factors, uncouples ERK/MAPK signaling from AXIN1 degradation, Wnt/β-catenin signal transduction and the coordinate regulation of apoptosis in these cell lines.
AXIN1 Plays a Central Role in Mediating Melanoma Cell Apoptosis with BRAF Inhibition
Previous in vitro studies using extracts derived from Xenopus oocytes elegantly recreated the cytoplasmic steps of the current Wnt/β-catenin pathway derived from genetic studies in model organisms, and also facilitated the development of kinetic modeling of the critical regulation of β-catenin stability by AXIN1 and APC.Citation25,Citation26 These studies pinpointed cellular abundance of AXIN1 as limiting step in the regulation of cellular β-catenin abundance by the so-called destruction complex comprised of AXIN1, APC and GSK3B, so it may not be all that surprising that AXIN1 abundance is a potential regulatory nexus of the crosstalk between Wnt/β-catenin and ERK/MAPK signaling. It will be critical to identify the mechanism by which ERK/MAPK inhibitors significantly decrease AXIN1 abundance in some cell lines, while having little measurable effect in other cell lines. Our previous studies found that the proteasome inhibitor MG132 inhibits loss of AXIN1 protein due to WNT3A and ERK/MAPK pathway inhibitor treatment in A375 melanoma cells,Citation24 suggesting that the loss of AXIN1 following WNT3A and ERK/MAPK inhibitor treatment in selected cell lines is mediated by proteasomal degradation. Consistent with a post-translational mechanism, changes in AXIN1 abundance were not correlated with changes at the transcriptional level.Citation24 A number of post-translational modifications of AXIN1 have previously been shown to regulate poly-ubiquitination and proteasomal degradation of AXIN1. Finding the mechanism by which ERK/MAPK regulates AXIN1 proteasomal degradation may be a matter of identifying the changes in AXIN1 post-translational modification following ERK/MAPK pathway inhibition.
In the absence of a Wnt ligand, the kinase glycogen synthase kinase-3β (encoded by GSK3B) phosphorylates AXIN1, promoting its stability.Citation27,Citation28 We have observed this phenomenon in apoptosis-sensitive melanoma lines (see ). In the presence of WNT3A, AXIN1 becomes dephosphorylatedCitation27 and degraded.Citation28 Pulse-chase analysis of cells following treatment with the GSK3B inhibitor lithium chloride (LiCl) suggested that GSK3B inhibition decreased AXIN1 phosphorylation and decreased AXIN1 half-life.Citation28 Conversely, treating cells with a broad-spectrum phosphatase inhibitor increased AXIN1 phosphorylation and AXIN1 half-life.Citation28 Furthermore, mutation of AXIN1 at putative GSK3B phosphorylation sites to phospho-dead alanine decreased the half-life of AXIN1 relative to wild-type.Citation27 The F-box class of ubiquitin ligases that regulate the proteasomal degradation of other components of the Wnt/β-catenin signaling pathway identify their substrates by phosphorylation status, and these reported observations suggest that AXIN1 may be similarly targeted for degradation by changes in phosphorylation.
Phosphorylation of AXIN1 by GSK3B is also regulated upstream by methylation. Protein arginine methyltransferase 1 (encoded by PRMT1) promotes the methylation of AXIN1 in the GSK3B-binding domain at arginine-378. This modification increases GSK3B association with AXIN1, increasing phosphorylation of AXIN1, decreasing ubiquitination and promoting stability.Citation29 Blocking ERK/MAPK signaling using a MEK inhibitor measurably changes the phosphorylation state of many proteins both directly and indirectly in melanoma and it will be important to determine whether the differential regulation of AXIN1 by some of these downstream ERK/MAPK-regulated effectors participates in determining the sensitivity of melanoma cell lines to apoptosis with inhibitors like vemurafenib.Citation30 In addition to phosphorylation and methylation, there is strong evidence that C terminal SUMOylation promotes the stability of AXIN1. Deleting the SUMOylation site on the six C terminal amino acids of AXIN1 decreases the half-life of AXIN1 relative to wild-type. Furthermore, restoring SUMOylation to the C terminus of AXIN1 by addition of a heterologous SUMOylation site decreases AXIN1 ubiquitination and restores stability of AXIN1.Citation31 Monitoring SUMOylation of AXIN1 following ERK/MAPK inhibition may offer insight into the mechanism by which ERK/MAPK signaling regulates AXIN1 stability.
AXIN1 stability is not only regulated by the cell prior to poly-ubiquitination, but is also regulated post-ubiquitination. RNF146 has been previously identified as an E3 ubiquitin ligase involved in PARsylation-dependent AXIN1 degradation, although its role downstream of ERK/MAPK inhibition remains as yet uncharacterized.Citation32,Citation33 As an endogenous counterbalance to AXIN1 degradation by PARsylation, de-phosphorylation, de-methylation and subsequent ubiquitination, the ubiquitin-specific protease USP34 has been shown to reverse ubiquitination of AXIN1, inhibiting its degradation. The transfection of cells with siRNA targeting USP34 increased AXIN1 ubiquitination, and decreased AXIN1 steady-state abundance.Citation34 It remains to be seen whether USP34 knockdown decreases AXIN1 in apoptosis-sensitive or apoptosis-resistant melanoma. However, a difference in USP expression or activity could conceivably regulate differences in response to BRAF inhibition between apoptosis-sensitive and apoptosis-resistant melanoma cell lines.
Translating Findings to Clinical Models of Melanoma
As with almost all melanoma research, one of the pressing questions is whether these findings have any potential impact on our diagnosis or treatment of patients with the disease. How does enhancement of apoptosis in cultured melanoma cell lines predict the actual response in a patient’s tumors? Apoptosis has been widely used as a surrogate benchmark for chemotherapeutic efficacy, and it’s inviting to imagine that certain patients with high levels of Wnt/β-catenin signaling (as measured by increased levels of nuclear β-catenin) may be more responsive to treatment with targeted BRAF inhibitors by virtue of enhanced cellular apoptosis. This prediction would also fit with the observation that apoptosis mediated by targeted BRAF inhibitors like PLX4720 requires β-catenin.Citation24 These studies also raise the question of whether activation of Wnt/β-catenin may have some therapeutic role in enhancing melanoma patient responses to targeted BRAF inhibition. Three large studies using independent tumor microarrays totaling over 1,000 patient tumors have associated increased levels of nuclear or cytosolic β-catenin in patient melanoma tumors to improved patient survival,Citation19,Citation22,Citation23 so consideration of Wnt/β-catenin activation as a therapeutic approach has already been broached.
Accumulating evidence suggests that any efforts to target Wnt/β-catenin signaling in melanoma (and likely in other cancers as well) will be complicated by the various effects of Wnt/β-catenin activation during different temporal stages of disease development. While patient data indicates that tumors with high levels of Wnt/β-catenin signaling have a more favorable clinical course, the interpretators of these data should keep in mind other studies suggesting a permissive role of Wnt/β-catenin in certain phases of melanoma progression, including melanoma development and metastasis. In transgenic mouse models, the melanocyte-specific expression of a constitutively-active, non-degradable, stabilized β-catenin mutant (β-catSTA, containing mutations at GSK3B phosphorylation target sites that normally target β-catenin for degradation) does not itself lead to increased spontaneous melanomas, but it does increase melanocyte immortalization through suppression of p16Ink4a (encoded in humans by CDKN2A) and also cooperates with melanocyte-specific mutant Nras to promote melanoma at a much higher frequency and rate.Citation35 Melanocyte-specific β-catSTA also enhances metastatic spread of melanoma in the presence of combined mutations in Pten (leading to activation of PI3K/AKT signaling) and Braf.Citation36 How are these results applicable to patient tumors that have already undergone the process of either transformation or metastasis? The answer to this important question will require more studies on how pathway crosstalk between Wnt/β-catenin, ERK/MAPK and PI3K/AKT signaling evolves throughout melanoma progression. With continued advances in targeted therapeutics and the mutational analysis of patient tumors, understanding the functional consequences of this pathway crosstalk will be essential for developing effective therapeutics that are tailored to individual patients.
Concluding Remarks
These studies, as well as those in our previously published manuscript,Citation24 have led to some surprising insight on how the interaction between different signaling pathways can impact the response of melanoma cells to emerging clinical therapies targeting BRAF and MEK signaling. Studies suggest that activation of Wnt/β-catenin signaling in melanoma through mutations in known canonical pathway members is probably rare.Citation37-Citation39 Nevertheless, clinical data has supported an important role for the association of this pathway’s activation state with changes in patient outcomes. Our observation that this pathway is required for melanoma cell apoptosis with targeted inhibition of the ERK/MAPK signaling axis was quite unexpected, and reminds us of the complexity of how signaling networks interact to regulate melanoma cell biology. Also surprising was our finding that AXIN1, a protein previously not implicated as a major regulator of melanoma biology, could play a key role in mediating both the crosstalk between Wnt/β-catenin signaling and act as a determinant of the cellular response to targeted ERK/MAPK inhibition.
Accumulating literature has also implicated a wide array of molecular mechanisms downstream of oncogenic BRAF mutations that may also be affected by the crosstalk with Wnt/β-catenin signaling. For example, BRAFV600E mutation in melanoma cells can lead to significant changes in epigenetic profiles,Citation40 particularly given the established association between β-catenin and chromatin modifying complexes.Citation41 Furthermore, the demonstrated crosstalk in melanoma cells between BRAF/MAPK signaling and metabolically responsive pathways such as AMPK/mTOR signaling, which regulates cellular proliferation and survival, may also involve Wnt/β-catenin signaling at some level.Citation42,Citation43 Clearly, more studies are needed to investigate how these findings may impact ongoing clinical efforts to target ERK/MAPK signaling in patients with metastatic melanoma.Citation44
Acknowledgments
The authors thank Mr. Jeffrey D. Lebowski for administrative assistance with preparation of the manuscript. Funding: W.H.C. is funded in part through a training grant from NIH (T.L.B. is funded in part through a training grant from NIH/NIAMS (T32AR056969). R.T.M. is an Investigator of the Howard Hughes Medical Institute. R.D.S. is a Medical Research Fellow of the Howard Hughes Medical Institute. We are indebted to these funding agencies for their continued support of our work. The contents of this manuscript are the sole responsibility of the authors and do not necessarily represent the official views of the NIAMS, NCI, NIH or the Howard Hughes Medical Institute.
Related Research Data
References
- Miller AJ, Mihm MC Jr.. Melanoma. N Engl J Med 2006; 355:51 - 65; http://dx.doi.org/10.1056/NEJMra052166; PMID: 16822996
- Cancer Facts & Figures. 2011.
- Kohler BA, Ward E, McCarthy BJ, Schymura MJ, Ries LAG, Eheman C, et al. Annual report to the nation on the status of cancer, 1975-2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst 2011; 103:714 - 36; http://dx.doi.org/10.1093/jnci/djr077; PMID: 21454908
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010; 467:596 - 9; http://dx.doi.org/10.1038/nature09454; PMID: 20823850
- Lee JT, Li L, Brafford PA, van den Eijnden M, Halloran MB, Sproesser K, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res 2010; 23:820 - 7; http://dx.doi.org/10.1111/j.1755-148X.2010.00763.x; PMID: 20973932
- Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA 2008; 105:3041 - 6; http://dx.doi.org/10.1073/pnas.0711741105; PMID: 18287029
- Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res 2010; 70:5518 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-10-0646; PMID: 20551065
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, et al. The Catalogue of Somatic Mutations in Cancer (COSMIC). Current Protocols in Human Genetics / Editorial Board, Jonathan L Haines [et Al] 2008; Chapter 10:Unit 10.1-Unit. 1.
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2010; 39:945 - 50
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al, BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507 - 16; http://dx.doi.org/10.1056/NEJMoa1103782; PMID: 21639808
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809 - 19; http://dx.doi.org/10.1056/NEJMoa1002011; PMID: 20818844
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012; 366:707 - 14; http://dx.doi.org/10.1056/NEJMoa1112302; PMID: 22356324
- Chien AJ, Conrad WH, Moon RT. A Wnt survival guide: from flies to human disease. J Invest Dermatol 2009; 129:1614 - 27; http://dx.doi.org/10.1038/jid.2008.445; PMID: 19177135
- Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998; 396:370 - 3; http://dx.doi.org/10.1038/24620; PMID: 9845073
- Dorsky RI, Moon RT, Raible DW. Environmental signals and cell fate specification in premigratory neural crest. Bioessays 2000; 22:708 - 16; http://dx.doi.org/10.1002/1521-1878(200008)22:8<708::AID-BIES4>3.0.CO;2-N; PMID: 10918301
- Dorsky RI, Raible DW, Moon RT. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev 2000; 14:158 - 62; PMID: 10652270
- Larue L, Kumasaka M, Goding CR. Beta-catenin in the melanocyte lineage. Pigment Cell Res 2003; 16:312 - 7; http://dx.doi.org/10.1034/j.1600-0749.2003.00050.x; PMID: 12753406
- Vance KW, Goding CR. The transcription network regulating melanocyte development and melanoma. Pigment Cell Res 2004; 17:318 - 25; http://dx.doi.org/10.1111/j.1600-0749.2004.00164.x; PMID: 15250933
- Bachmann IM, Straume O, Puntervoll HE, Kalvenes MB, Akslen LA. Importance of P-cadherin, beta-catenin, and Wnt5a/frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin Cancer Res 2005; 11:8606 - 14; http://dx.doi.org/10.1158/1078-0432.CCR-05-0011; PMID: 16361544
- Kageshita T, Hamby CV, Ishihara T, Matsumoto K, Saida T, Ono T. Loss of beta-catenin expression associated with disease progression in malignant melanoma. Br J Dermatol 2001; 145:210 - 6; http://dx.doi.org/10.1046/j.1365-2133.2001.04336.x; PMID: 11531781
- Maelandsmo GM, Holm R, Nesland JM, Fodstad O, Flørenes VA. Reduced beta-catenin expression in the cytoplasm of advanced-stage superficial spreading malignant melanoma. Clin Cancer Res 2003; 9:3383 - 8; PMID: 12960126
- Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci USA 2009; 106:1193 - 8; http://dx.doi.org/10.1073/pnas.0811902106; PMID: 19144919
- Meyer S, Fuchs TJ, Bosserhoff AK, Hofstädter F, Pauer A, Roth V, et al. A seven-marker signature and clinical outcome in malignant melanoma: a large-scale tissue-microarray study with two independent patient cohorts. PLoS ONE 2012; 7:e38222; http://dx.doi.org/10.1371/journal.pone.0038222; PMID: 22685558
- Biechele TL, Kulikauskas RM, Toroni RA, Lucero OM, Swift RD, James RG, et al. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci Signal 2012; 5:ra3; http://dx.doi.org/10.1126/scisignal.2002274; PMID: 22234612
- Lee E, Salic A, Krüger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol 2003; 1:E10; http://dx.doi.org/10.1371/journal.pbio.0000010; PMID: 14551908
- Salic A, Lee E, Mayer L, Kirschner MW. Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell 2000; 5:523 - 32; http://dx.doi.org/10.1016/S1097-2765(00)80446-3; PMID: 10882137
- Willert K, Shibamoto S, Nusse R. Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes Dev 1999; 13:1768 - 73; http://dx.doi.org/10.1101/gad.13.14.1768; PMID: 10421629
- Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase-3β regulates its stability. J Biol Chem 1999; 274:10681 - 4; http://dx.doi.org/10.1074/jbc.274.16.10681; PMID: 10196136
- Cha B, Kim W, Kim YK, Hwang BN, Park SY, Yoon JW, et al. Methylation by protein arginine methyltransferase 1 increases stability of Axin, a negative regulator of Wnt signaling. Oncogene 2011; 30:2379 - 89; http://dx.doi.org/10.1038/onc.2010.610; PMID: 21242974
- Old WM, Shabb JB, Houel S, Wang H, Couts KL, Yen CY, et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell 2009; 34:115 - 31; http://dx.doi.org/10.1016/j.molcel.2009.03.007; PMID: 19362540
- Kim MJ, Chia IV, Costantini F. SUMOylation target sites at the C terminus protect Axin from ubiquitination and confer protein stability. FASEB J 2008; 22:3785 - 94; http://dx.doi.org/10.1096/fj.08-113910; PMID: 18632848
- Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN, et al. Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS ONE 2011; 6:e22595; http://dx.doi.org/10.1371/journal.pone.0022595; PMID: 21799911
- Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 2011; 13:623 - 9; http://dx.doi.org/10.1038/ncb2222; PMID: 21478859
- Lui TTH, Lacroix C, Ahmed SM, Goldenberg SJ, Leach CA, Daulat AM, et al. The ubiquitin-specific protease USP34 regulates axin stability and Wnt/β-catenin signaling. Mol Cell Biol 2011; 31:2053 - 65; http://dx.doi.org/10.1128/MCB.01094-10; PMID: 21383061
- Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M, et al. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev 2007; 21:2923 - 35; http://dx.doi.org/10.1101/gad.450107; PMID: 18006687
- Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, et al. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011; 20:741 - 54; http://dx.doi.org/10.1016/j.ccr.2011.10.030; PMID: 22172720
- Omholt K, Platz A, Ringborg U, Hansson J. Cytoplasmic and nuclear accumulation of beta-catenin is rarely caused by CTNNB1 exon 3 mutations in cutaneous malignant melanoma. Int J Cancer 2001; 92:839 - 42; http://dx.doi.org/10.1002/ijc.1270; PMID: 11351304
- Pollock PM, Hayward N. Mutations in exon 3 of the beta-catenin gene are rare in melanoma cell lines. Melanoma Res 2002; 12:183 - 6; http://dx.doi.org/10.1097/00008390-200204000-00013; PMID: 11930117
- Rimm DL, Caca K, Hu G, Harrison FB, Fearon ER. Frequent nuclear/cytoplasmic localization of beta-catenin without exon 3 mutations in malignant melanoma. Am J Pathol 1999; 154:325 - 9; http://dx.doi.org/10.1016/S0002-9440(10)65278-9; PMID: 10027390
- Hou P, Liu D, Dong J, Xing M. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle 2012; 11:286 - 95; http://dx.doi.org/10.4161/cc.11.2.18707; PMID: 22189819
- Genovese G, Ghosh P, Li H, Rettino A, Sioletic S, Cittadini A, et al. The tumor suppressor HINT1 regulates MITF and β-catenin transcriptional activity in melanoma cells. Cell Cycle 2012; 11:2206 - 15; http://dx.doi.org/10.4161/cc.20765; PMID: 22647378
- Namiki T, Coelho SG, Hearing VJ. NUAK2: an emerging acral melanoma oncogene. Oncotarget 2011; 2:695 - 704; PMID: 21911917
- Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, et al. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell 2009; 33:237 - 47; http://dx.doi.org/10.1016/j.molcel.2008.12.026; PMID: 19187764
- Corcoran RB, Settleman J, Engelman JA. Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget 2011; 2:336 - 46; PMID: 21505228