Abstract
Ultraviolet light (UV) can provoke genome instability, partly through its ability to induce homologous recombination (HR). However, the mechanism(s) of UV-induced recombination is poorly understood. Although double-strand breaks (DSBs) have been invoked, there is little evidence for their generation by UV. Alternatively, single-strand DNA lesions that stall replication forks could provoke recombination. Recent findings suggest efficient initiation of UV-induced recombination in G1 through processing of closely spaced single-strand lesions to DSBs. However, other scenarios are possible, since the recombination initiated in G1 can be completed in the following stages of the cell cycle. We developed a system that could address UV-induced recombination events that start and finish in G2 by manipulating the activity of the sister chromatid cohesion complex. Here we show that sister-chromatid cohesion suppresses UV-induced recombination events that are initiated and resolved in G2. By comparing recombination frequencies and survival between UV and ionizing radiation, we conclude that a substantial portion of UV-induced recombination occurs through DSBs. This notion is supported by a direct physical observation of UV-induced DSBs that are dependent on nucleotide excision repair. However, a significant role of nonDSB intermediates in UV-induced recombination cannot be excluded.
Keywords: :
Genetic Recombination
Genetic recombination is a universal DNA transaction that is important both for increasing genetic diversity in meiosis and for maintaining genome integrity. Years ago it was suggested that recombination is initiated by double-strand breaks (DSBs).Citation1 Indeed, a DSB caused by a site-specific endonuclease is sufficient to cause homologous recombination (HR) that can be detected both physically and genetically (for example, see refs. Citation2 and Citation3). Following DSB induction of random or site-specific DSBs (summarized in ref. Citation4), preferential strand resection at the ends leads to the exposure of 3′ single-strand DNA (ssDNA). Subsequently, the ssDNA invades the homologous molecule, and DNA synthesis extends the joint molecule. Finally, the joint molecule is resolved (; for a review of DSB HR see refs. Citation5 and Citation6). All of these steps can be detected in real-time.Citation3,Citation4 While some key biological processes such as meiosis and mating-type switching use endonuclease-induced DSBs to drive homologous recombination, most mitotic recombination is likely to stem from spontaneous lesions that may resemble the many kinds of DNA damaging agents that can induce high frequencies of recombination.Citation7-Citation10 Ionizing radiation (IR) can cause DSBs as well as single-strand (ss) breaks. UV can induce recombination; however, it does not cause significant amounts of DSB, at least not directly.Citation11 Importantly, formation of DSBs may not be essential to induce homologous recombination. Early recombination models were based on ss breaks, not DSBs, as initiating events.Citation6,Citation12-Citation14 Later studies confirmed ss breaks as inducers of recombination (for example, see ref. Citation15).
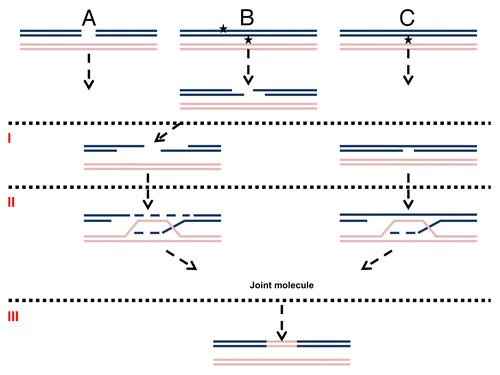
Figure 1. Mechanisms of DNA damage induced recombination. (A) Recombination induced by a direct DSB. (B) Recombination induced by a secondary (indirect) DSB. (C) Recombination induced by gaps or nicks. The first stage in homologous recombination is processing of the primary lesion (I). For direct DSBs (A) it is resection. Adjacent lesions on opposite strands (represented as stars) are first converted into a DSB and then resected (B). Nicks or gaps (C) may be expanded without forming a DSB. The second step (II) is strand invasion, DNA synthesis and creation of a joint molecule. The third step (III) is resolution of homologous intermediates (for simplicity only one possibility of resolution is presented).
UV Lesions and UV-Induced Recombination
In the budding yeast Saccharomyces cerevisiae, UV can induce recombination between sister chromatids as well as homologous or heterologous chromosomes.Citation8,Citation16 A recent study of UV-induced loss of heterozygosity (LOH) by the Petes labCitation17 suggested that UV, a well-known mutagen, is more efficient at inducing recombination between homologous chromosomes than base substitutions on a per lesion base. Unlike for endonucleases or IR,Citation4,Citation18 the opportunity to follow UV-induced recombination in real-time is limited. The major UV-induced DNA lesions consist of cyclobutane pyrimidine dimers and 4–6 photoproducts. Under conditions used to examine UV-induced recombinationCitation19,Citation20 the incidence of lesions is typically around 0.1–1 per Kb. However, closely opposed lesions are formed at high doses.Citation21,Citation22 The lesions are efficiently repaired by nucleotide excision repair (NER) (see refs. Citation23–Citation25 and references therein). Intermediates in NER include nicks and short gaps, which have been proposed as initiators of recombination (see below) either directly or in relation to stalled replication forks.Citation8,Citation26,Citation27 The nicks and gaps created by NER could be recombinogenic with or without being processed to DSBs (). Lesions that escape NER until replication would appear in single-strand regions created by the replicative DNA helicase. The lesions in ssDNA would not be subject to repair and could block the replication fork. The resulting gapped DNA at stalled forks or subsequent DSBs could be rescued through homologous recombination.Citation8,Citation26
UV-Induced Recombination at G1 Suggests A DSB Intermediate
Allelic recombination (recombination between homologous chromosomes) can be an important contributor to genome instability. It often results in loss of heterozygosity (LOH), which is a major mechanism for inactivation of WT alleles in disease as well as asexual evolution. By using a unique genetic system where cells could not enter S phase unless allelic recombination had occurred, it was shown that UV-irradiation of G1 cells can induce allelic recombination before cells enter S phase.Citation28 Yet, it is possible that most UV-induced recombination events are initiated during DNA replication even when the damage is inflicted in G1. Recently, this issue was elegantly tackled by the Petes lab.Citation17 Through an examination of the pattern of UV-induced LOH, they deduced how and when recombination is triggered (). They concluded that for cells irradiated at G1, recombination is also initiated in G1 (or before the damaged locus is replicated) and occurs through a DSB mechanism.Citation17 Their approach involved first the identification of daughter colonies using a color-based, colony-sectoring assay. Then, they addressed LOH across the genome by following 55,000 polymorphic sites of heterozygosity. Genome-wide analysis of LOH enabled them to decipher the recombinational fate of the four sister chromatids ().Citation17
Figure 2. Analysis of UV-induced LOH for cells irradiated at G1. Cells that are heterozygous at a specific locus (“a” allele and “c” allele) are arrested at G1 and irradiated with UV (damage is represented by a star). Sectored colonies are selected after UV exposure.Citation17 Each sector subcolony represents two sister chromatids. Therefore, the four chromatids are represented in the two sector colonies. Sequencing each sector can reveal which allele, “a” or “c,” was lost. If the number of sequence reads for “a” and “c” alleles is equal then heterozygosity is maintained. If there are no sequence reads for one of them, LOH has occurred. Through analysis of LOH in the two sectors it is possible to establish a 3:1 or 4:0 ratio for the four sister chromatids (see text and ref. Citation17).
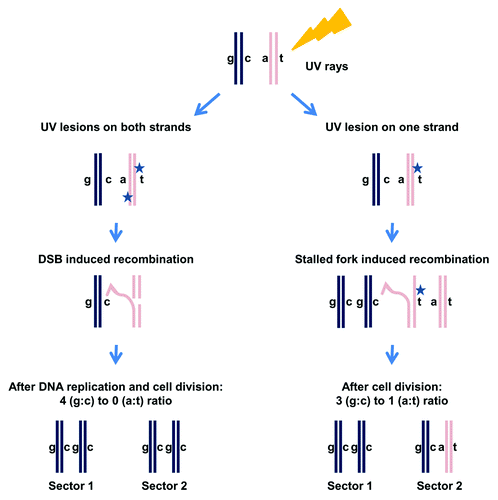
Although DNA damage was induced in G1 (before DNA replication),Citation17 the subsequent recombination could be initiated during replication of the damaged locus or even after replication. With the Petes system, it is possible to address the type of lesion that provokes recombination as well the cell cycle stage in which recombination occurs. One model suggests that a UV lesion on one strand is converted to a DSB during DNA replication. It predicts that LOH events should give rise to three chromatids with the same sequence at the site of recombination and one chromatid that bears a different sequence (the chromatid that emerged from the undamaged strand) (). A 3:1 ratio is also characteristic of LOH generated in G2-arrested cells from DSBs directly induced by ionizing radiation (IR);Citation29 nevertheless, other scenarios that do not involve DSB can also give a 3:1 ratio. However, if closely spaced UV lesions on opposite strands cause DSBs before DNA replicationCitation30 ( and , left side), and they are repaired using information from the homologous chromosome, there will be no sequence heterozygosity in the daughter chromatids, resulting in a 4:0 ratio for LOH. The results from the Petes studyCitation17 are consistent with UV leading to DSBs in G1 (or before replication), such that subsequent repair after cells enter S-phase results in a 4:0 LOH pattern.
UV Initiation and Completion of Recombination is Revealed in Yeast with Decreased Levels of Cohesin
While the generation of DSBs by closely opposed UV lesions in G1 conveniently explains the observed 4:0 LOH, there are alternative explanations that involve gaps or nicks especially for gene conversion events without associated crossovers. For example, sequential repair of two UV lesions found on opposite strands but distant from each other may result in a 4:0 ratio. A recent study shows that reduction of dNTP pools in G1 cells leads to delayed gap filling during NER; consequently, the initial gap can be extended by Exo1.Citation31 A 4:0 ratio can be achieved if the gap generated by attempts to repair the first UV lesion is repaired by homologous recombination, and if the DNA that was synthesized during the recombination then serves as a template for the gap filling stage of the repair of the second UV lesion. Moreover, there may be other alternative explanations to the 4:0 outcome, since repair is completed at a different stage of the cell cycle after passing the replication start point.Citation32 For example, unrepaired lesions on opposing strands of a duplex could interfere with replication of both the leading and lagging strands. A 4:0 ratio could occur if the synthesis on one strand were rescued by the homologous chromosome with or without break formation and the other used it as a template.
Although the mechanisms of UV-induced recombination at G1 and G2 may differ, the findings with UV-induced LOH in G1 cells and the possible roles of replication could be addressed with a system in which UV-induced recombination between homologous chromosomes is initiated and completed in G2. Measuring recombination at G2 would exclude the complexities associated with a subsequent round of replication. However, damage-induced allelic recombination is low in G2.Citation7,Citation33 Recently, we found that by manipulating the levels of cohesin complex by changing the gene dosage of the cohesin gene MCD1 it is possible to greatly increase recombination induced by DNA damage administered to G2 cells.Citation33 We established that damage-induced recombination between homologs is suppressed by cohesin in mitotic cells () using an inter-homolog recombination assay. The same assay is used in and ; the strains contain 5′ and 3′ truncated alleles of TYR1 with an overlap of 400 nucleotides. Recombination between the homologous chromosomes within the TYR1 locus results in the ability to grow on media lacking Tyrosine. Recombination frequencies are determined by the number of TYR+ colonies divided by total survivors; for details, see ref. Citation33. The induced recombinants/106 survivors correspond to frequency of events after DNA damage treatment less the no treatment control frequency. We found that reduction of the Mcd1 component within the sister chromatid cohesion complex resulted in a 10–20-fold increase in both UV- and IR-induced allelic recombination when cells are irradiated at G2, whereas there was only an approximate 2-fold increase in recombination for cells irradiated at G1Citation33 (). Importantly, the survival of WT tetraploid and MCD1 simplex cells exposed to 20 krad in G2 is similar (80% vs. 50%, respectively), and there is no difference in survival after 40 J/m2 UV exposure. Therefore, the difference in inter-homolog recombination cannot be attributed to any large effect on survival. These observations suggest that UV-induced allelic recombination can be very efficient in G2, if sister chromatid cohesion is compromised. Hence, passage of cells though S phase is not required for recombination.
Figure 3. Altering cohesin affects damage-induced recombination. (A) Cohesin channels recombination toward sister chromatids and away from homologous chromosomes in G2. This model describes the role of cohesin in preventing homologous recombination (see ref. Citation33; the reproduction of the model was done according to the Creative Commons Attribution License -2.5 of PloS). (B) Tetraploid WT and the hypomorphic “simplex” cohesion mutant with one copy of MCD1 (for details see text and ref. Citation33) were irradiated with IR or UV in the G1 or G2 phases of the cell cycle. Presented are the frequencies of DNA damage-induced allelic recombination per unit radiation. For the x-axis, the unit dose was J/m2 for UV and krad for IR. Symbols: WT G1, white solid squares; WT G2, white striped squares; MCD1 simplex G1, gray solid squares; MCD1 simplex G2, gray striped squares. (C) Four scenarios that utilize temperature changes to alter cohesin activity in the mcd1–1 temperature-sensitive mutant are presented: 23◦C, light green; 30◦ C, red. In Scenarios 1, 2 and 4, the logarithmic growth of the culture and the G2 arrest occur at the same temperature. In Scenario 3, the cells are grown to logarithmic phase at 30◦ C and arrested with nocodazole for 90 min at the same temperature, but then the temperature is shifted to 23◦ C for 30 min before irradiation while remaining in nocadazole. (D) Recombination induced by 20 krad IR or 40 J/m2 for each Scenario in (C).
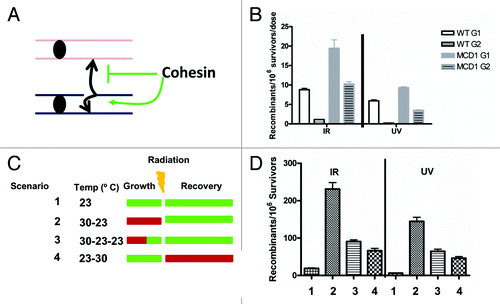
Figure 4. Significant contribution of DSBs to UV-induced recombination. (A) Survival curve of rad52-null cells arrested in G2 by nocodazole and treated with IR. The survival of rad52-null cells treated with 40 J/m2 is placed on the trend-line. The vertical gray line represents the dose in krad equivalent to 40 J/m2. (B) Dose-dependent IR-induced recombination is presented for two mcd1 mutants; MCD1 simplex (solid line) and mcd1–1 (dashed line). The data for mcd1–1 is taken from , Scenario 2 and for MCD1 simplex from . Recombination induced by 40 J/m2 was calculated for both MCD1 mutants and placed on the trend-line. Vertical gray lines represent the dose in krad equivalent to 40 J/m2. (C) Detection of DSB formation after UVB and ionizing radiation are as previously reported.Citation43,Citation44 Briefly, overnight cultures of WT, mcd1–1 and rad14 haploid strains were grown at 30°C, arrested with nocododazole and then exposed to 40J/m2 UVB in ice-cold water. The cultures were then returned to YPDA containing nocodazole to incubate for up to 4 h (30°C). As a control, cells were irradiated with IR (5,10,20,40 krad) using a 137Cs irradiator and harvested immediately after irradiation. Cells were processed for PFGE analysis as described.Citation45 Linear chromosomes III (details in text) were determined by Southern blotting with a CHA1 probe; the zone corresponding to linear chromosome III or its derivatives is marked by a rectangle. The solid arrow indicates a resected linear DNA molecule and the dashed arrow points to a possible linear dimer of chromosome III (details in text). (D) IR dose-dependent Southern blot signal from a chromosome III specific probe [represented as AU (Arbitrary Units)]. Gray solid and Gray empty circles represent the chromosome III southern signals obtained for WT and mcd1–1 cells irradiated with 40 J/m2 after 4 h, respectively (see lane 11 and 17 of panel 4C).
![Figure 4. Significant contribution of DSBs to UV-induced recombination. (A) Survival curve of rad52-null cells arrested in G2 by nocodazole and treated with IR. The survival of rad52-null cells treated with 40 J/m2 is placed on the trend-line. The vertical gray line represents the dose in krad equivalent to 40 J/m2. (B) Dose-dependent IR-induced recombination is presented for two mcd1 mutants; MCD1 simplex (solid line) and mcd1–1 (dashed line). The data for mcd1–1 is taken from Figure 3B, Scenario 2 and for MCD1 simplex from Figure 3A. Recombination induced by 40 J/m2 was calculated for both MCD1 mutants and placed on the trend-line. Vertical gray lines represent the dose in krad equivalent to 40 J/m2. (C) Detection of DSB formation after UVB and ionizing radiation are as previously reported.Citation43,Citation44 Briefly, overnight cultures of WT, mcd1–1 and rad14 haploid strains were grown at 30°C, arrested with nocododazole and then exposed to 40J/m2 UVB in ice-cold water. The cultures were then returned to YPDA containing nocodazole to incubate for up to 4 h (30°C). As a control, cells were irradiated with IR (5,10,20,40 krad) using a 137Cs irradiator and harvested immediately after irradiation. Cells were processed for PFGE analysis as described.Citation45 Linear chromosomes III (details in text) were determined by Southern blotting with a CHA1 probe; the zone corresponding to linear chromosome III or its derivatives is marked by a rectangle. The solid arrow indicates a resected linear DNA molecule and the dashed arrow points to a possible linear dimer of chromosome III (details in text). (D) IR dose-dependent Southern blot signal from a chromosome III specific probe [represented as AU (Arbitrary Units)]. Gray solid and Gray empty circles represent the chromosome III southern signals obtained for WT and mcd1–1 cells irradiated with 40 J/m2 after 4 h, respectively (see lane 11 and 17 of panel 4C).](/cms/asset/215a8c14-f1bb-450b-b7f9-ec1ff5c01717/kccy_a_10921945_f0004.gif)
While the experiments with reduced levels of cohesin provided an approach to addressing events initiated in G2, the extent to which events occurring in G2 contributed to recombination was not clear. To address directly UV-induced recombination within G2 (i.e., before mitosis), we have utilized temperature-sensitive mcd1 diploid mutants, since cohesin activity could be adjusted simply by varying the temperature at different stages of the cell cycle. Although cohesin is required for recombinational repair of DSBs in G2, cells are capable of repair at a semi-permissive temperature.Citation33
Overnight cultures grown at permissive temperature (23°C) were diluted and grown to logarithmic phase at permissive (green in ), or semi-permissive temperature, 30°C (red in ) for 3 h. Subsequently the cells were arrested in G2 with nocodazole for 2 more hours at the same temperature. Cells were then irradiated with UV or IR and spread to plates that were incubated at permissive or semi-permissive temperatures for recovery and colony formation. In Scenario 1 (), cells were grown, arrested and incubated following irradiation at permissive temperature. Under these conditions, DNA damage-induced recombination was low. In Scenario 2, cells were grown to logarithmic phase and arrested by nocodazole at semi-permissive temperature, irradiated and subsequently incubated at the permissive temperature. If recombination occurs within G2, allelic recombination should be high, since cohesin is partially inactive, allowing greater interaction between homologous chromosomes. However, if recombination occurs outside of G2 (i.e., after cell division and/or requires replication), the reduced cohesin activity at semi-permissive temperature should not make a difference, since Mcd1 is degraded during anaphase and is highly expressed in early S phase.Citation34,Citation35 We found that allelic recombination was 10-fold higher for Scenario 2 as compared with Scenario 1. Therefore, recombination induced by exposure of G2 cells to UV or IR appears to be confined to G2 (). Under these conditions, there is no significant difference in the survival between Scenario 1 and Scenario 2 (40% vs. 30%, respectively, for 20 krad, p = 0.1; 57 ± 3 vs. 49 ± 5, respectively, for 40J/m2)
When cells were grown and arrested at semi-permissive temperature but shifted to permissive temperature 30 min before irradiation while still in nocodazole (Scenario 3, “30-23-23”), the level of induced-recombination was reduced ~2-fold as compared with Scenario 2 but still higher than Scenario 1. Therefore, despite the shift to permissive temperature, sister chromatid cohesion capacity is not fully restored. This is not surprising, since while cohesin can be synthesized in G2, it has to be activated in order to be cohesive. Usually, activation occurs in S phase and not in G2. However, DNA damage can activate cohesin,Citation36,Citation37 even outside of S phase. Yet, the role of DNA damage-induced cohesion in recombination is not clear.Citation38, Citation39 Using the conditions of Scenario 3 the DNA damage reactivation of sister chromatid cohesion and its ability to suppress UV- and IR-induced allelic recombination could be studied by manipulating genes that control DNA damage-induced cohesion. Finally, for cells grown and arrested at permissive temperature, irradiated at G2 and recovered at semi-permissive temperature (Scenario 4) there was a 3–5-fold increase in DNA damaged-induced allelic recombination compared with cells kept only at the permissive temperature (Scenario 1). However, the frequencies were ~3-fold less than for cells grown at semi-permissive temperature and recovered at permissive temperature (Scenario 2). Importantly, under the conditions of Scenario 4, there is a reduction in survival in response to DNA damage comparing to Scenario 1 (permissive temperature) (20% vs. 40%, respectively, for IR and about 20% vs. 60%, respectively, for UV). Therefore, it is not clear what the direct effect on recombination is and what is the indirect effect due to reduction of survival.
Collectively, these results suggest that manipulation of temperature results in rapid changes in Mcd1 activity in the mcd1-1 temperature-sensitive mutant. As a result, it is possible to alter quickly the choice between sister chromatid (i.e., suppression of homologous recombination) and homologous chromosomes. Based on the results from the different scenarios, we conclude that cohesin has to be active at the time of DNA damage induction or shortly after. The increase in allelic recombination under Scenario 4 vs. Scenario 1 can be explained in two ways. First, it could be that some UV or IR DNA lesions are converted to DSBs during DNA replication at the next cell cycle when cohesin is already compromised, leading to increased allelic recombination.Citation40 Second, the events may still occur in G2. Some recombination events can occur a long time after induction of DNA damage while the cells are still at G2Citation33,Citation41 and, for the case of Scenario 4, after cohesin activity is reduced.
UV-Induced Cell Death and Recombination Frequencies Suggest Both DSB and Non-DSB Intermediates
While these results with the mcd1 mutants support the view of a replication-independent mechanism for UV-induced recombination, there remains the question of whether DSBs might be involved. We suggest that UV-derived DSBs can contribute significantly, although probably not solely, to the UV-induced recombination. Since DSBs are lethal in rad52-deficient cells, the survival response of rad52 cells treated with ionizing radiation could be used to estimate the number of the proposed UV-derived DSBs. For rad52-null cells irradiated in G2, a 40 J/m2 UV dose was comparable to survival after 5 krad (). If much of the UV-induced killing were due to DSBs, then 40 J/m2 would yield an amount of DSBs corresponding to 5 krad. (In fact, under these conditions not all the lethality events in rad52-deficient cells are due to un-repaired DSB; some of them are due to other DNA lesions; therefore, the IR equivalent dose may be lower than 5 krad.) However, as determined for two very different mcd1 mutants (mcd1-1 and MCD1 simplex), the recombination frequency induced by 40 J/m2 UV in mcd1 mutants was equivalent to 13 krad IR ( and and ref. Citation33). These data suggest that UV induces recombination more effectively than killing rad52 cells. If UV can lead to DSBs, then they would account for only around 40% (5/13) of the UV-induced recombination events.
We examined DSB induction directly using an assay we developed that is based on linearization of circular chromosome III.Citation4,Citation42-Citation44 Presented in are results from an initial set of experiments in which cells were irradiated with 40 J/m2, and the chromosomes were displayed using pulse-field gel electrophoresis (PFGE) (precise evaluations are the subject of future investigations). As expected, there were no linear chromosome III molecules immediately following UV-irradiation. However, they did appear at 1 h, and by 4 h there was a shift in position. This “PFGE shift” is likely due to resection at the ends of the broken DNA molecules (marked with a solid arrow on ), as described by Westmoreland et al.Citation4 Higher molecular weight forms of chromosome III (dashed arrows ) are in the size range that would correspond to a linear dimer of chromosome III, which is likely due to recombination.Citation4 Similar results were obtained for mcd1–1 cells. Interestingly, the formation of the resected chromosome III was highly dependent on the activity of NER. Linear size chromosome III molecules did not appear in a rad14-deleted strain (), demonstrating that the NER mutation rad14 may prevent break accumulation induced by UV when cells are irradiated in G2.
These PFGE determined results are consistent with the view in that excision repair could lead to overlapping gaps, which would be seen as DSBs, similar to our findings with methyl-methanesulfonate (MMS).Citation42,Citation43,Citation45 Based on the direct induction of DSBs by IR, it appears that the amount of linearized molecules resulting from NER after 40 J/m2 is in the range of that produced by 1–5 krad or around 5 to 10 DSBs/cell ( and ref. Citation4). The nature and mode of formation of these breaks is under current investigation. Regardless, the impact of a NER mutation might be expected to reduce the lethal effects of UV in rad51 and rad52-null cells;this expectation is in contrast to the synergistic interaction between NER mutants and recombination mutants when treated with UV Citation46. The apparent contradiction can be easily resolved; although there would be no DSBs associated with removal of UV-lesions in the NER-deficient cells, many more DSBs might arise upon subsequent entry of cells into S phase due the blockage of replication by the enormous amounts of unrepaired lesions. In the absence of the recombination machinery, both replication fork collapse and increased UV mutagenesis can lead to cell death.
Overall, we present evidence that supports DSB intermediates in UV-induced recombination but does not exclude the possibility of other intermediates. Quantitative measurements of UV-induced recombination in G2 cells using different assays are needed to assess the extent to which lesions other than DSBs contribute to the recombination. Using the system we described to regulate cohesin function during the cell cycle () provides the opportunity to identify additional genes involved in various types of DNA damage-induced recombination, including UV. The possibility that UV can induce recombination at G2 and in a DSB-independent manner has interesting implications regarding the mechanism by which cohesin controls HR. So far, cohesion was thought to control HR through recruitment to DSBs.Citation47,Citation48 It remains to be seen if cohesin can be recruited to other recombination intermediates. Of particular interest, and a topic currently under study, is the role of cohesin molecules deposited during normal S phase on allelic recombination.
Concluding Remarks
The skin of humans can experience as many as 100,000 UV lesions per dayCitation49 (and references therein) some of which could lead to LOH. Since LOH can change the balance between WT and mutant alleles, it is important to understand mechanisms of UV-induced recombination, especially the genetic risk for increased LOH. Since cohesin properties in humans are similar to yeast, the approaches described here are particularly relevant to addressing the role that cohesin may play in protecting the human genome and preventing disease associated with genome instability.
Acknowledgments
We thank Drs. Steve Roberts and Kin Chan for critically evaluating this manuscript. This work was supported by the Intramural Research Program of the NIEHS (NIH, DHHS) under project 1Z01ES065073 to M.A.R.
Related Research Data
References
- Resnick MA. The repair of double-strand breaks in DNA; a model involving recombination. J Theor Biol 1976; 59:97 - 106; http://dx.doi.org/10.1016/S0022-5193(76)80025-2; PMID: 940351
- Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 2010; 329:82 - 5; http://dx.doi.org/10.1126/science.1191125; PMID: 20595613
- Hicks WM, Yamaguchi M, Haber JE. Real-time analysis of double-strand DNA break repair by homologous recombination. Proc Natl Acad Sci USA 2011; 108:3108 - 15; http://dx.doi.org/10.1073/pnas.1019660108; PMID: 21292986
- Westmoreland J, Ma W, Yan Y, Van Hulle K, Malkova A, Resnick MA. RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma-induced double-strand break ends. PLoS Genet 2009; 5:e1000656; http://dx.doi.org/10.1371/journal.pgen.1000656; PMID: 19763170
- Andersen SL, Sekelsky J. Meiotic versus mitotic recombination: two different routes for double-strand break repair: the different functions of meiotic versus mitotic DSB repair are reflected in different pathway usage and different outcomes. Bioessays 2010; 32:1058 - 66; http://dx.doi.org/10.1002/bies.201000087; PMID: 20967781
- Haber JE, Ira G, Malkova A, Sugawara N. Repairing a double-strand chromosome break by homologous recombination: revisiting Robin Holliday’s model. Philos Trans R Soc Lond B Biol Sci 2004; 359:79 - 86; http://dx.doi.org/10.1098/rstb.2003.1367; PMID: 15065659
- Kadyk LC, Hartwell LH. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 1992; 132:387 - 402; PMID: 1427035
- Kadyk LC, Hartwell LH. Replication-dependent sister chromatid recombination in rad1 mutants of Saccharomyces cerevisiae. Genetics 1993; 133:469 - 87; PMID: 8454200
- Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol 2005; 25:7158 - 69; http://dx.doi.org/10.1128/MCB.25.16.7158-7169.2005; PMID: 16055725
- Fasullo M, Dong Z, Sun M, Zeng L. Saccharomyces cerevisiae RAD53 (CHK2) but not CHK1 is required for double-strand break-initiated SCE and DNA damage-associated SCE after exposure to X rays and chemical agents. DNA Repair (Amst) 2005; 4:1240 - 51; http://dx.doi.org/10.1016/j.dnarep.2005.06.006; PMID: 16039914
- Rapp A, Greulich KO. After double-strand break induction by UV-A, homologous recombination and nonhomologous end joining cooperate at the same DSB if both systems are available. J Cell Sci 2004; 117:4935 - 45; http://dx.doi.org/10.1242/jcs.01355; PMID: 15367581
- Meselson MS, Radding CM. A general model for genetic recombination. Proc Natl Acad Sci USA 1975; 72:358 - 61; http://dx.doi.org/10.1073/pnas.72.1.358; PMID: 1054510
- Holliday R. The recombination, repair and modification of DNA. DNA Repair (Amst) 2011; 10:993 - 9; http://dx.doi.org/10.1016/j.dnarep.2011.04.005; PMID: 22066132
- Haber JE. Evolution of Models of Homologous Recombination. In: Lankenau REaD-H, ed. Recombination and meiosis. Berlin: Springer-Verlag, 2008:1-64.
- Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell 2004; 117:171 - 84; http://dx.doi.org/10.1016/S0092-8674(04)00301-0; PMID: 15084256
- Fasullo M, Sun M. UV but not X rays stimulate homologous recombination between sister chromatids and homologs in a Saccharomyces cerevisiae mec1 (ATR) hypomorphic mutant. Mutat Res 2008; 648:73 - 81; http://dx.doi.org/10.1016/j.mrfmmm.2008.09.009; PMID: 18929581
- St Charles J, Hazkani-Covo E, Yin Y, Andersen SL, Dietrich FS, Greenwell PW, et al. High-resolution genome-wide analysis of irradiated (UV and γ-rays) diploid yeast cells reveals a high frequency of genomic loss of heterozygosity (LOH) events. Genetics 2012; 190:1267 - 84; http://dx.doi.org/10.1534/genetics.111.137927; PMID: 22267500
- Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci USA 2008; 105:11845 - 50; http://dx.doi.org/10.1073/pnas.0804529105; PMID: 18701715
- Resnick MA, Setlow JK. Repair of pyrimidine dimer damage induced in yeast by ultraviolet light. J Bacteriol 1972; 109:979 - 86; PMID: 4551759
- Wheatcroft R, Cox BS, Haynes RH. Repair of UV-induced DNA damage and survival in yeast. I. Dimer excision. Mutat Res 1975; 30:209 - 18; PMID: 1107831
- Resnick MA, Westmoreland J, Amaya E, Bloom K. UV-induced damage and repair in centromere DNA of yeast. Mol Gen Genet 1987; 210:16 - 22; http://dx.doi.org/10.1007/BF00337753; PMID: 3323836
- Lam LH, Reynolds RJ. A sensitive, enzymatic assay for the detection of closely opposed cyclobutyl pyrimidine dimers induced in human diploid fibroblasts. Mutat Res 1986; 166:187 - 98; http://dx.doi.org/10.1016/0167-8817(86)90017-9; PMID: 2429177
- Franklin WA, Doetsch PW, Haseltine WA. Structural determination of the ultraviolet light-induced thymine-cytosine pyrimidine-pyrimidone (6-4) photoproduct. Nucleic Acids Res 1985; 13:5317 - 25; http://dx.doi.org/10.1093/nar/13.14.5317; PMID: 4022781
- Setlow RB. Cyclobutane-type pyrimidine dimers in polynucleotides. Science 1966; 153:379 - 86; http://dx.doi.org/10.1126/science.153.3734.379; PMID: 5328566
- E.C FRIEDBERG GCW. W. SIEDE, R. D. WOOD, R. A. SCHULTZ DNA Repair and Mutagenesis. Washington DC.: ASM press, 2006.
- Galli A, Schiestl RH. Cell division transforms mutagenic lesions into deletion-recombinagenic lesions in yeast cells. Mutat Res 1999; 429:13 - 26; http://dx.doi.org/10.1016/S0027-5107(99)00097-4; PMID: 10434021
- Galli A, Schiestl RH. On the mechanism of UV and gamma-ray-induced intrachromosomal recombination in yeast cells synchronized in different stages of the cell cycle. Mol Gen Genet 1995; 248:301 - 10; http://dx.doi.org/10.1007/BF02191597; PMID: 7565592
- Fabre F. Induced intragenic recombination in yeast can occur during the G1 mitotic phase. Nature 1978; 272:795 - 8; http://dx.doi.org/10.1038/272795a0; PMID: 347306
- Lee PS, Petes TD. From the Cover: mitotic gene conversion events induced in G1-synchronized yeast cells by gamma rays are similar to spontaneous conversion events. Proc Natl Acad Sci USA 2010; 107:7383 - 8; http://dx.doi.org/10.1073/pnas.1001940107; PMID: 20231456
- Reynolds RJ. Induction and repair of closely opposed pyrimidine dimers in Saccharomyces cerevisiae. Mutat Res 1987; 184:197 - 207; http://dx.doi.org/10.1016/0167-8817(87)90017-4; PMID: 2444878
- Giannattasio M, Follonier C, Tourrière H, Puddu F, Lazzaro F, Pasero P, et al. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell 2010; 40:50 - 62; http://dx.doi.org/10.1016/j.molcel.2010.09.004; PMID: 20932474
- Symington LS. Initiation and completion of spontaneous mitotic recombination occur in different cell cycle phases. Proc Natl Acad Sci USA 2010; 107:8045 - 6; http://dx.doi.org/10.1073/pnas.1003050107; PMID: 20418501
- Covo S, Westmoreland JW, Gordenin DA, Resnick MA. Cohesin Is limiting for the suppression of DNA damage-induced recombination between homologous chromosomes. PLoS Genet 2010; 6:e1001006; http://dx.doi.org/10.1371/journal.pgen.1001006; PMID: 20617204
- Xiong B, Gerton JL. Regulators of the cohesin network. Annu Rev Biochem 2010; 79:131 - 53; http://dx.doi.org/10.1146/annurev-biochem-061708-092640; PMID: 20331362
- Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 1997; 91:47 - 57; http://dx.doi.org/10.1016/S0092-8674(01)80008-8; PMID: 9335334
- Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, et al. A molecular determinant for the establishment of sister chromatid cohesion. Science 2008; 321:566 - 9; http://dx.doi.org/10.1126/science.1157880; PMID: 18653894
- Ström L, Karlsson C, Lindroos HB, Wedahl S, Katou Y, Shirahige K, et al. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 2007; 317:242 - 5; http://dx.doi.org/10.1126/science.1140649; PMID: 17626884
- Ström L. Additional proof for the importance of Eco1 for DNA double-strand break repair. Cell Cycle 2010; 9:3644; http://dx.doi.org/10.4161/cc.9.18.13182; PMID: 20930507
- Sjögren C, Ström L. S-phase and DNA damage activated establishment of sister chromatid cohesion--importance for DNA repair. Exp Cell Res 2010; 316:1445 - 53; http://dx.doi.org/10.1016/j.yexcr.2009.12.018; PMID: 20043905
- Cortés-Ledesma F, Aguilera A. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep 2006; 7:919 - 26; http://dx.doi.org/10.1038/sj.embor.7400774; PMID: 16888651
- Covo S, Westmoreland JW, Reddy AK, Gordenin DA, Resnick MA. RAD53 is limiting in double-strand break repair and in protection against toxicity associated with ribonucleotide reductase inhibition. DNA Repair (Amst) 2012; 11:317 - 23; http://dx.doi.org/10.1016/j.dnarep.2011.12.008; PMID: 22277748
- Ma W, Panduri V, Sterling JF, Van Houten B, Gordenin DA, Resnick MA. The transition of closely opposed lesions to double-strand breaks during long-patch base excision repair is prevented by the coordinated action of DNA polymerase delta and Rad27/Fen1. Mol Cell Biol 2009; 29:1212 - 21; http://dx.doi.org/10.1128/MCB.01499-08; PMID: 19075004
- Ma W, Westmoreland JW, Gordenin DA, Resnick MA. Alkylation base damage is converted into repairable double-strand breaks and complex intermediates in G2 cells lacking AP endonuclease. PLoS Genet 2011; 7:e1002059; http://dx.doi.org/10.1371/journal.pgen.1002059; PMID: 21552545
- Ma W, Westmoreland J, Nakai W, Malkova A, Resnick MA. Characterizing resection at random and unique chromosome double-strand breaks and telomere ends. Methods Mol Biol 2011; 745:15 - 31; http://dx.doi.org/10.1007/978-1-61779-129-1_2; PMID: 21660686
- Ma W, Resnick MA, Gordenin DA. Apn1 and Apn2 endonucleases prevent accumulation of repair-associated DNA breaks in budding yeast as revealed by direct chromosomal analysis. Nucleic Acids Res 2008; 36:1836 - 46; http://dx.doi.org/10.1093/nar/gkm1148; PMID: 18267974
- Game JC, Cox BS. Synergistic interactions between rad mutations in yeast. Mutat Res 1973; 20:35 - 44; http://dx.doi.org/10.1016/0027-5107(73)90095-X; PMID: 4586553
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, et al. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell 2004; 16:991 - 1002; http://dx.doi.org/10.1016/j.molcel.2004.11.027; PMID: 15610741
- Ström L, Lindroos HB, Shirahige K, Sjögren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell 2004; 16:1003 - 15; http://dx.doi.org/10.1016/j.molcel.2004.11.026; PMID: 15610742
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179 - 204; http://dx.doi.org/10.1016/j.molcel.2010.09.019; PMID: 20965415