Abstract
Cellular inhibitor of apoptosis proteins (cIAP1 and cIAP2) function to prevent apoptosis and are often overexpressed in various cancers. However, mutations in cIAP1/2 can activate the alternative NFκB pathway through IκBα-kinase-α (IKKα) and are associated with hematopoetic malignancies. In the current study, we found that knockdown of cIAP2 in human mammary epithelial cells resulted in activation of MDM2 through increased SUMOylation and profound reduction of the pool of MDM2 not phosphorylated at Ser166. cIAP2 siRNA markedly decreased p53 levels, which were rescued by addition of the MDM2 inhibitor, Nutlin3a. An IAP antagonist, which induces cIAP degradation, transiently increased MDM2 mRNA. Simultaneous transfection of siRNA for cIAP2 and IKKα reduced MDM2 protein, while expression of a kinase-dead IKKβ strongly increased non-Ser166 P-MDM2. Inhibition of either IKKα or -β partially rescued p53 levels, while concomitant IKKα/β inhibition fully rescued p53 after cIAP2 knockdown. Surprisingly, IKKα knockdown alone increased SUMO-MDM2, suggesting that in the absence of activation, IKKα can prevent MDM2 SUMOylation. cIAP2 knockdown disrupted the interaction between the MDM2 SUMO ligase, PIAS1 and IKKα. Partial knockdown of cIAP2 cooperated with V12H-ras-transfected mammary epithelial cells to enhance colony formation. In summary, our data identify a novel role for cIAP2 in maintaining wild-type p53 levels by preventing both an NFκB-mediated increase and IKKα/-β-dependent transcriptional and post-translational modifications of MDM2. Thus, mutations or reductions in cIAP2 could contribute to cancer promotion, in part, through downregulation of p53.
Introduction
Proteins of the inhibitor of apoptosis protein (IAP) gene family have emerged as among the most important intrinsic inhibitors of apoptosis. They are characterized by the presence of a baculovirus IAP repeat (BIR) domain in one to three copies. Two cIAP members cIAP1 and -2 function as ubiquitin ligases for caspase-3 and -7.Citation1,Citation2 cIAP1 and -2 are also required for NFκB signaling from the activated tumor necrosis factor receptor-1 (TNFR1) by mediating the ubiquitination of RIP1.Citation3-Citation5 Both cIAP1 and -2 mediate K48-ubiquitination of the NFκB-inducing kinase NIK,Citation6 resulting in NIK proteasomal degradation, thereby preventing NFκB activation in unstimulated cells.Citation5-Citation7
The involvement of the IAPs in cancer is underscored by the frequent induction of these genes in several tumor types.Citation8 cIAP1 is an established oncogene seen in DNA amplicons (with cIAP2) in mice and human tumors, and can cooperate in transformation with YAP1Citation9 or with Myc.Citation10 Oncogenic activity of both cIAP1/2 can be attributed both to their anti-apoptotic role and their involvement TNF-mediated NFκB activation. As such, ongoing efforts directed at reducing cIAP levels in cancers are underway.Citation8,Citation11-Citation13 Reduction of both cIAP1 and -2 by small-molecule IAP antagonists results in stabilization and activation of NIK, leading to engagement of the alternative NFκB pathway and autocrine production of TNFα in some cells to result in cell death.Citation5,Citation7
NFκB transcription factors play an important role in many cancers.Citation14,Citation15 There are two IκB kinases (IKKs) that activate NFκB: IKKα and IKKβ, as well as a regulatory subunit, IKKγ (NEMO). IKKβ preferentially phosphorylates the IκB proteins, resulting in their ubiquitination and degradation, followed by the release and activation of canonical NFκB p50/p65 (RelA) and c-Rel/p65 complexes. IKKα can be specifically activated by the NFκB-inducing kinase, NIK.Citation16 IKKα homodimer-mediated phosphorylation of cytoplasmic NFκB2/p100 results in partial proteasomal processing of p100 to produce mature p52, thereby activating the alternative pathway.Citation17
The tumor suppressor p53 exerts governance over cellular responses to genotoxic stress, wherein it mediates a variety of outcomes including cell cycle arrest, senescence and apoptosis. Cellular levels of p53 are regulated, in large part, through the action of the E3-ubiquitin ligase, MDM2 (murine double minute 2). MDM2 is transcriptionally regulated by p53 and is subject to numerous posttranslational modifications. Among these, phosphorylation at Ser166 and Ser186 facilitate nuclear entry,Citation18 p300 bindingCitation19 and enhance E3-ligase activity.Citation20,Citation21 Nuclear MDM2 can be SUMOylated, which is mediated in part by PIAS proteins.Citation22 SUMOylation of MDM2 prevents autoubiquitination, resulting in MDM2 stabilization and p53-directed ubiquitination, while a SUMO-specific protease that removes SUMO from MDM2 stabilizes p53.Citation23
Importantly, NFκB can negatively regulate p53. Specifically, reconstitution of IKKβ, but not IKKα, was shown to increase Mdm2 levels and decrease p53 in mouse IKKα/-β−/− mouse embryo fibroblasts (MEFs). The effects of IKKβ in these cells were inhibited by coexpression of a dominant-negative IκBα super repressor which suggests that canonical NFκB was responsible for regulating Mdm2 levels.Citation24
Given the potential importance of the cIAPs as targets in cancer therapy and the known effect of IAP antagonism in activating NFκB, we sought to determine whether reduction of cIAP2 alone would induce NFκB and impact on p53 levels. Using cIAP2 siRNA, shRNA and IAP antagonists, we demonstrate that knockdown (KD) of cIAP2 using several methods downregulates p53 protein in both an IKKα- and IKKβ-dependent manner. Moreover, we uncover novel functions for both these kinases in regulating posttranslational modifications of MDM2. Lastly, we extended these findings by demonstrating that reduction or ablation of cIAP2 increases V12H-ras-induced colony formation.
Results
cIAP2 protein expression regulates p53 levels
Previous studies have demonstrated that IKKβ can regulate p53 protein stability by increasing MDM2 expression through the canonical NFκB pathway.Citation24 Since IAP antagonists reduce the cIAPs in conjunction with the activation of IKKα, we wished to determine if cIAP2 KD alone (1) would activate NFκB and (2) alter p53 protein expression. MCF-10AT1 cells (wild-type p53 MCF-10A human mammary epithelial cells transfected with V12H-rasCitation26) were transfected with cIAP2 siRNA. shows that downregulation of cIAP2 alone resulted in a marked reduction in p53. Knockdown of cIAP1 weakly reduced p53 levels; however, this was accompanied by strong stabilization of cIAP2, likely a function of loss of cIAP1-mediated polyubiquitination of cIAP2.Citation27 Following transfection of both siRNAs, cIAP2 was again stabilized; however, a strong reduction in p53 was observed. The interaction between p53 and MDM2, and subsequent ubiquitination and degradation, is regulated in part through phosphorylation of p53 at Ser15/20 and other phosphoacceptor sites. Moreover, phosphorylation at these residues promotes the transcriptional activity of p53.Citation28,Citation29 To explore whether cIAP2 downregulation alone would impact on p53 phosphorylation, we performed immunoblotting for phospho-p53 Ser15 [P-p53(Ser15)]. Densitometry showed that while p53 was reduced by approximately 45% relative to control after cIAP downregulation, P-p53 (Ser15) was reduced by 60%, indicating a small net decrease in phosphorylation. Overall, however, the major effect of cIAP2 KD was on p53 levels, not its phosphorylation. shows that p21 levels were strongly decreased in association with cIAP2 KD and reduction of p53.
Figure 1. Downregulation of cIAPs in mammary epithelial cells reduces p53. (A) MCF-10AT1 cells were either non-transfected or transfected with non-targeting siRNA (NT) or cIAP1 and cIAP2 siRNAs, as indicated, for 48 h. Ten μg of cell lysate was immunoblotted for cIAP1 and -2 (top panel with non-anti-rat IAP (RIAP) antibody that recognizes both cIAP1 and -2, cIAP2 (Epitomics antibody), phospho-p53 (Ser15) and total p53. The asterisk indicates a non-specific immunoreactive band. (B) MCF-10AT1 cells transfected with non-targeting (NT) siRNA or cIAP2 siRNA for 48 h were immunoblotted for cIAP2, p53 and p21. (C) MCF-10AT1 cells were incubated with 50 or 100 nM AEG40730 for 6 h. Control cells were treated with vehicle for 12 h. Ten μg of cell lysate was subjected to immunoblotting for cIAP1/2 (RIAP), cIAP2, phospho-p53 (Ser15) and total p53. (D) MCF-10AT1 cells were exposed to vehicle (DMSO) or SM164 (5 nM) for 4 and 8 h. Immunoblotting was performed for cIAP2, total p53, phospho-IKKα/-β and total IKKα. (E) MCF-10AT1 cells were transfected with XIAP siRNA and lysates isolated after 48 h in culture and subjected to immunoblotting for XIAP, cIAP2 and p53. (F) MCF-10AT1 cells were infected with an adenovirus to overexpress human cIAP2 or a control LacZ adenovirus. Immunoblotting was performed on lysates 48 h following infection for P-IKKα, IKKα and cIAP2. Actin immunoreactivity was used as a loading control on all blots. Images shown are representative of independent experiments performed a minimum of three times.
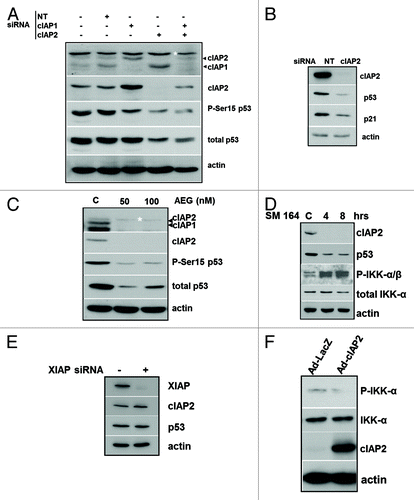
IAP antagonists simultaneously reduce cIAP1 and cIAP2 proteins.Citation30 The IAP antagonists SM164Citation30 and AEG40730Citation4 both reduced cIAP1 and cIAP2 and markedly reduced p53 in MCF-10AT1 cells as well as P-p53(Ser15) (). Ablation of cIAP1 and -2 with SM164 also increased IKKα and -β phosphorylation (). Bivalent SMAC mimetics such as SM164 have been shown to block XIAP interaction with caspases at higher concentrations.Citation31 To test the effect of blocking XIAP function on p53, we transfected MCF-10AT1 cells with a XIAP siRNA. p53 remained unchanged after KD of XIAP in , providing evidence that XIAP does not play a role in p53 regulation following IAP antagonist treatment. To see if overexpression of cIAP2 would have the reciprocal effect, we infected cells with a cIAP2 adenoviral expression vector or control adenovirus. The basal levels of phosphorylated IKKα were clearly reduced in cIAP2-overexpressing cells, consistent with the reciprocal ability of high levels of cIAP2 to block IKK activity ().
Taken together, these data demonstrate that KD of either of the cIAPs, but in particular cIAP2, using RNAi and IAP antagonists, activates IKKs and negatively regulates p53 expression.
cIAP2 knockdown induces MDM2 Ser166 phosphorylation and SUMOylation to repress p53
Since MDM2 is the predominant E3 ligase that regulates p53, we first determined whether MDM2 was involved in the reduction of p53 in cIAP2-depleted cells. cIAP1 or cIAP2 siRNA-transfected MCF-10AT1 cells were treated with Nutlin-3a, which prevents MDM2-mediated ubiquitination.Citation32 Nutlin-3a completely rescued p53 from downregulation when cIAP1 or -2 were reduced (). The MDM2 protein was also stabilized, since autoubiquitination is also blocked by Nutlin-3a. Thus, MDM2 activity is required for cIAP2 depletion induced p53 downregulation.
Figure 2. cIAP2 depletion results in posttranslational modifications of MDM2 (A) MCF-10AT1 cells were transfected with either a non-targeting (NT) or cIAP2 siRNA for 48 h as described in Materials and Methods. Some lanes contain protein lysate from cells incubated for 3 h prior to harvesting with Nutlin-3a (10 μM). Fifteen μg of cell lysate was subjected to immunoblotting for p53, MDM2 and cIAP1/2. (B) MCF-10AT1 cells were transfected with cIAP2 or NT siRNA as above. Some cells were treated with actinomycin D (ActD) (2 μg/mL) for the final 3 h. Antibodies for immunoblotting were p53, MDM2 (SMP14), which recognizes 95 kDa MDM2, P-MDM2 (Ser166) and cIAP2. (C) MCF-10AT1 cells were transfected with siRNA for cIAP2 or NT as indicated. Cells were then treated with MG132 or vehicle (DMSO) for the final 2 h. Lysates were immunoblotted for MDM2 (SMP14) and P-MDM2(Ser166). (D) MCF-10AT1 cells were transfected with either NT or cIAP2 siRNA as indicated. RNA was extracted 48 h later and qRT-PCR was performed to assess levels of MDM2 mRNA. (E) MCF-10AT1 cells were treated with SM164 for the indicated times. RNA was extracted and qRT-PCR was performed to assess levels of MDM2 mRNA and expressed as fold difference compared with vehicle (DMSO) treated cells. (F) Cells were transfected with cIAP2 siRNA or NT in the presence or absence of an expression construct for Flag-tagged IκBαSR and harvested after 48 h of culture. Protein lysates were used for immunoblotting as shown. The latter experiment was performed once. All other experiments were independently repeated three times. In all immunoblot experiments, actin was used as a protein loading control.
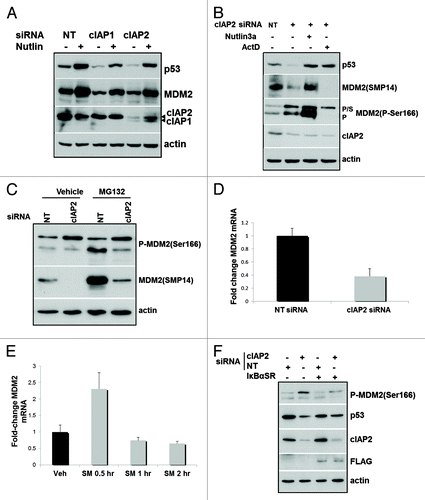
Ser166 phosphorylation promotes MDM2 nuclear translocation and interaction with p53;Citation18 however, the SMP14 antibody employed does not recognize MDM2 that has been phosphorylated at Ser166.Citation33 Using a P-MDM2(Ser166) antibody, shows that cIAP2 siRNA transfection resulted in an increase in the total P-MDM2 (Ser166) detected at both 95 kDa and 120 kDa compared with cells treated with the non-targeting siRNA. SUMO-1-conjugated MDM2 directly interacts with p53Citation34 and has an apparent molecular weight of 120 kDa.Citation23,Citation35 In our experiments, we only detected the 120 kDa MDM2 with anti-P-MDM2 (Ser166) and not SMP14, consistent with a requirement for Ser166 phosphorylation for SUMOylation. Again, Nutlin-3a stabilized both p53 and MDM2 following cIAP2 KD. Next, cIAP2 siRNA-transfected cells were treated with actinomycin D for 3 h prior to harvest to prevent de novo transcription. This treatment completely eliminated both non-P-Ser166 MDM2 and the 95 kDa P-MDM2 (Ser166) but had little effect on the SUMOylated form of MDM2. In summary, these data show that cIAP2 KD induces an increase in phosphorylation of MDM2 at Ser166 and SUMOylation of the phosphorylated protein. Once SUMOylated, the MDM2 protein is highly stabilized.
Although cIAP2 KD increased P-MDM2 (Ser166) and SUMO-MDM2, the level of SMP14-reactive MDM2 was strongly reduced (). This was surprising, given that previous studies have shown that NFκB positively regulates MDM2 transcription.Citation24 To assess if this was a result of reduced MDM2 stability, we treated cells for the final 2 h of culture with the proteasome inhibitor, MG132, after transfection with non-targeting (NT) or cIAP2 siRNA. shows that, as expected, MG132 stabilized both the Ser166 phosphorylated and non-phosphorylated forms of 95 kDa MDM2 but had no effect on the 120 kDa SUMOylated form. In contrast, MG132 had little effect on P-MDM2 (Ser166) when added to cIAP2 KD cells relative to cIAP2 KD alone and produced only a small increase in SMP14-reactive MDM2. The reduction in MDM2 protein following cIAP2 KD was therefore not the result of destabilization.
We next performed qRT-PCR to look for effects on MDM2 transcripts. A strong decrease in MDM2 transcripts was observed 48 h following siRNA-mediated cIAP2 KD (), likely subsequent to the concurrent low levels of p53. In order to look at earlier time points after loss of cIAP2, we performed qRT-PCR after treatment with SM164 for short periods. shows that MDM2 mRNA levels transiently increased immediately following reduction of the cIAPs and then were rapidly reduced. To confirm that NFκB activation was required for the induction of MDM2 transcription after cIAP2 KD, we transiently transfected cells with the mutant IκBα (IκBα super repressor mutated at Ser 32/36), which blocks canonical NFκB activation. shows that inhibition of canonical NFκB strongly reduced MDM2 after cIAP2 KD.
Overall, these data indicate that cIAP2 KD transiently induces MDM2 transcription, in an NFκB-dependent manner followed by its phosphorylation at Ser166 and SUMOylation to mediate p53 reduction. Thereafter, MDM2 transcripts are decreased, correlating with the reduction in p53.
IKKα regulates MDM2 SUMOylation while IKKβ regulates MDM2 Ser166 phosphorylation
We next assessed the roles of IKKα and -β in MDM2 and p53 regulation following cIAP2 KD. IKKα siRNA was transfected into MCF-10AT1 cells in the presence of non-targeting or cIAP2 siRNA. shows that the sole KD of IKKα reduced SMP14-reactive MDM2. Moreover, we observed an increase in the 120 kDa SUMO-MDM2 associated with a reduction in p53 protein. Inhibition of IKKβ activity by adenoviral expression of the IKKβKA Kinase decreased both forms of P-MDM2 (Ser166) while markedly increasing the level of SMP14-reactive MDM2 (compare lanes 1 and 3). Thus, IKKβ appears to constitutively promote Ser166 phosphorylation of MDM2, although its activity is not necessary to maintain MDM2 protein under basal conditions. Predictably, p53 levels remained unchanged. Consistent with a requirement for Ser166 phosphorylation prior to SUMOylation, the simultaneous IKKα ablation and IKKβ inhibition resulted in an overall decrease in P-MDM2 (Ser166) as well as the accumulation of SMP14-reactive MDM2 without a detectable change in the level of p53.
Figure 3. IKKα and IKKβ are both required for MDM2 activation and p53 downregulation following cIAP2 knockdown. MCF-10AT1 cells were transfected with cIAP2 or IKKα siRNA then infected with an adenovirus expressing a dominant-negative mutant of IKKβ. Non-targeting siRNA and adenovirus expressing GFP were used as transfection and infection controls. Protein extracts were immunoblotted for p53, P-p53 (Ser15), MDM2 (SMP14), P-MDM2 (Ser166), IKKβ, IKKα and cIAP2. The immunoblot shown is representative of three separate experiments.
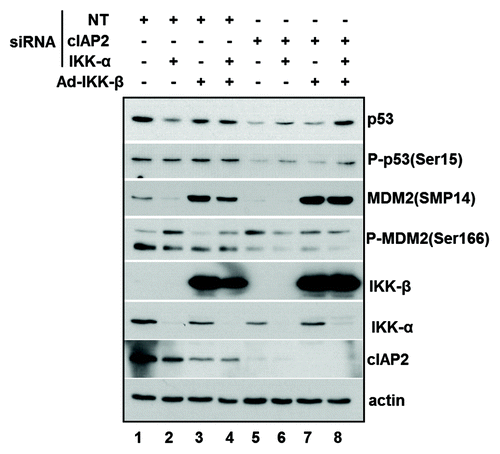
We next examined the effects of IKK inhibition in the context of cIAP2 KD. While a low basal level of NFκB activity is present in MCF-10AT1 cells, cIAP2 KD activates both IKKα and -β. As in , cIAP2 siRNA transfection resulted in an increase in the 120kDa P-Ser166/SUMO-MDM2 form (, compare lanes 1 and 5) while the level of SMP14-reactive MDM2 was sharply reduced. Co-transfection of siRNA for both IKKα and cIAP2 (lane 6) resulted in a reduction of both forms of P-MDM2 (Ser166) along with a similar loss of SMP14-reactive MDM2 seen after cIAP2 KD alone. Consistent with this, the reduction of postranslationally modified MDM2 was accompanied by a partial rescue of p53. Two separate IKKα siRNAs also partially rescued p53 after cIAP2 KD (Fig. S1). Inhibition of IKKβ activity in cIAP2-depleted cells again reduced P-MDM2 (Ser166) and resulted in a large increase in the steady-state levels of SMP14-reactive MDM2. The reduction in P-MDM2 (Ser166) was also associated with partial rescue of p53, which can be attributed to the requirement for Ser166 phosphorylation for activation of MDM2.Citation36 Simultaneous inhibition of both kinases in the presence of cIAP2 siRNA (lane 7) produced a response with elements of the individual effects, reducing both 120 kDa and 95 kDa-P-MDM2 (Ser166), increasing levels of SMP14-reactive MDM2 and fully rescuing p53.
These results combined with those in demonstrate that negative regulation of p53 associated with cIAP2 downregulation is dependent on signaling to MDM2 by both IKKα and -β. IKKα activation following cIAP2 KD can increase MDM2 protein levels through activation of IKKβ and the canonical pathway. While IKKβ appears to constitutively promote Ser166 phosphorylation of MDM2, this is further increased following cIAP2 KD and IKK activation. Both the activation and absence of IKKα appear to be permissive for MDM2 SUMOylation suggesting that IKKα has a non-catalytic function in tonically repressing MDM2 SUMOylation.
Loss of cIAP2 releases SUMO ligase PIAS1 from IKKα
The SUMO-E3 ligase PIAS1 can mediate SUMOylation of p53 to inhibit its activity.Citation37 PIAS1 and other E3 ligases can also SUMOylate MDM2, which increases its ubiquitin ligase activity toward p53.Citation41 Interestingly, PIAS1 and IKKα have been shown to form a complex in macrophages. IKKα-mediated phosphorylation of PIAS1 at Ser90 disrupts this complex.Citation38 Thus, we reasoned that cIAP2 KD-associated IKKα activation would similarly release PIAS1, which might then SUMOylate MDM2. To explore this possibility, MCF-10AT1 cells were transfected with NT or cIAP2 siRNA and subjected to reciprocal immunoprecipitation (IP) and immunoblot of endogenous protein with anti-PIAS1 or anti-IKKα. demonstrates that PIAS1 and IKKα were present in IKKα and PIAS1 IPs, respectively, in control cells. Strikingly, this interaction was drastically inhibited by cIAP2 KD. Immunoblotting confirmed the cIAP2 KD in lysates of the cIAP2 siRNA-transfected cells used for IP and indicated that cIAP2 was not present in the IKKα:PIAS1 complex. In , non-immune IgG IPs confirmed the specificity of the IP antibodies. Thus, loss of cIAP2 and accompanying activation of IKKα disrupts its interaction with PIAS1.
Figure 4. The IKKα interaction with PIAS1 SUMO ligase is disrupted following cIAP2 downregulation. (A) MCF-10AT1 cells were transfected with cIAP2 or NT siRNAs and 250 μg of protein were immunoprecipitated with anti-IKKα or anti-PIAS1. Complexes were separated by SDS-PAGE and immunoblotted with anti-IKKα or anti-PIAS1. Input was 5% of lysate used for IP and was also reacted with anti-cIAP2 to monitor KD. (B) To assess specificity of the IPs, lysate from NTsiRNA-transfected cells was immunoprecipitated with non-immune IgG and immunoreactivity compared with input lysate by immunoblot with anti-IKKα or anti-PIAS1. (C) MCF-10AT1 cells were transfected with NT, cIAP2 and PIAS1 siRNA as indicated. Whole-cell lysates were immunoblotted for PIAS1, P-MDM2 (Ser166) and MDM2 (SMP14). Note that cell lysates were run on the same gel but one lane was moved next to a non-adjacent lane as indicated for clarity. The asterisks in (A and B) indicate a higher mol wt anti-IKKα cross-reactive band. Actin was used as a loading control. Images shown are representative of two independent experiments.
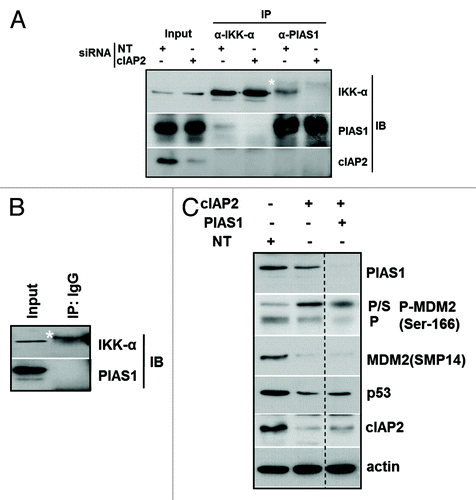
To assess the role of PIAS1 in SUMOylation of MDM2 following KD of cIAP2, we co-transfected PIAS1 siRNA into MCF-10AT1 cells in the presence of cIAP2 siRNA. The immunoblots in show that KD of cIAP2 again induced both phosphorylated forms of MDM2; however, although KD of both cIAP2 and PIAS1 resulted in a strong decrease in the 95kDa P-MDM2 (Ser166), SUMO-MDM2 underwent a smaller reduction relative to cIAP2 KD alone. As expected, p53 levels were reduced following cIAP2 KD, while transfection of siRNA for PIAS1 along with cIAP2 partially rescued p53 levels. Taken together, these results suggest that either IKKα KD and or its activation following cIAP2 KD may allow SUMOylation of MDM2, at least in part, through a mechanism involving disruption of a complex containing PIAS1 and IKKα.
Stable cotransfection of cIAP2 shRNA activates IKKα and cooperates with V12H-ras transformation
Since IAP mutation has been associated with activation of NFκB and correlated with some cancers, we next investigated the effect of cIAP2 depletion following transformation with V12H-ras. Cells transfected with activated Ras can stabilize and activate wild-type p53 to result in senescence.Citation39 We therefore reasoned that p53 downregulation in cIAP2-depleted cells might facilitate transformation. To test this, MCF-10A cells were cotransfected with V12H-ras and either control non-targeting or cIAP2 shRNA containing plasmids. Stably transfected cells were selected in G418 and clones were isolated and expanded. As shown in , cIAP2 shRNA expressing cells formed greater numbers of colonies relative to control clones when cotransfected with V12H-ras. The expression of V12H-ras was confirmed by immunoblot analysis of isolated colonies (). Vector-transfected cells (C) are also shown. All MCF-10A (V12H-ras/NT shRNA) and (V12H-ras/cIAP2 shRNA) clones expressed on average twice the level of H-ras relative to vector control cells, indicative of similar transfection efficiencies between colonies. Comparison of V12H-ras transfected cells with vector-transfected MCF-10A cells showed that the expression of cIAP2 was strongly increased in V12H-ras-transfected cells, consistent with a previous report.Citation40 Although p53 levels were not different in V12H-ras expressing cells relative to vector-transfected cells, p53 Ser15 phosphorylation was increased. Transfection of V12H-ras and cIAP2 shRNA in isolated colonies resulted in an average decrease in cIAP2 protein of approximately 40% in cIAP2 relative to control MCF-10A (V12H-ras/NT shRNA). This partial KD cIAP2 may be due in part to the strong counterinduction of cIAP2 by both V12H-ras and NFκB signaling and is similar to the reduction of cIAP2 achieved by others with a different cIAP2 targeted duplex.Citation41 However, even stable partial reduction of cIAP2 expression consistently resulted in reduced p53/P-p53 (Ser15). Downregulation of cIAP2 also resulted in an increase in both total and phosphorylated NIK, consistent with the role of cIAP2 as a ubiquitin ligase for NIK, targeting it for degradation.Citation6 The DNA damage-induced protein GADD45α is negatively regulated by NFκB and positively regulated by p53.Citation42 Immunoblotting with anti-GADD45α demonstrated a strong reduction of this protein in cells expressing cIAP2 shRNA. cIAP2 shRNA had no effect on cIAP1 protein levels, indicating that the observed reduction in p53 was attributable to the reduced cellular levels of cIAP2 protein.
Figure 5. Reduction in cIAP2 protein levels in vitro increases colony formation in V12H-ras-expressing human mammary epithelial cells, activates IKKα and canonical NFκB. MCF-10A human mammary epithelial cells were cotransfected with V12H-ras with either cIAP2 shRNA or a control shRNA or transfected with vector only (pcDNA3). (A) Colonies were selected in G418 in triplicate and stained in 0.2% crystal violet for enumeration and results presented for cells transfected with the indicated plasmids. Bars represent mean ± SD of triplicate plates. (B) Immunoblot analysis of protein lysates derived from stable clones of MCF-10A cells transfected with V12H-ras + control shRNA or V12H-ras + cIAP2 shRNA. Lysates were immunoblotted for H-ras, cIAP2, NIK, P-NIK, p53, P-p53 (Ser15) and GADD45α. Densitometry is shown for cIAP2 protein. (C) Stable clones were immunoblotted for phospho-IKKα/β and total IKKα and IKKβ. Actin was used as a control for protein loading. (D) Nuclear extracts from the indicated stable clones were subjected to EMSA using an NFκB oligonucleotide as a probe. Supershift analysis was performed using anti-p50 antibody. Shifted and supershifted complexes are indicated. (E) The same nuclear extracts shifted on probe for Sp1 as a loading control. Images shown are representative of two independent experiments.
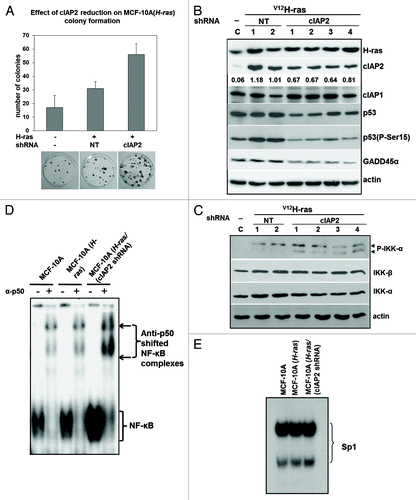
Since cIAP downregulation has been shown to activate NFκB through stabilization of NIK,Citation5,Citation7 we next assessed activation of IKKs using phospho-IKK antibodies. V12H-ras expression alone induced IKKβ phosphorylation (P-IKKβ); however, stable KD of cIAP2 resulted in the differential appearance of P-IKKα (). No change in the expression of either IKKα or IKKβ was observed. To assess NFκB, we performed EMSA using a canonical NFκB DNA probe on nuclear extracts from two pooled V12H-ras/NTshRNA clones and three pooled V12H-ras/cIAP2 shRNA clones. shows that V12H-ras expression in MCF-10A cells produced a small induction of NFκB activity; however, NFκB was markedly increased in cells expressing cIAP2 shRNA. Supershift analysis indicated that DNA-binding activity was entirely attributable to canonical p50/NFκB1 as either a homodimer (lower complex) or heterodimer (upper complex). Replicate samples of the same nuclear extracts were used to shift an SP1 probe to provide evidence of equivalent loading (). Thus, cIAP2 downregulation activates the canonical NFκB pathway in V12H-ras-transformed cells.
Together, these results show that partial reduction in cIAP2 is sufficient to activate NFκB and this activation is associated with enhanced V12H-ras-induced colony formation.
Discussion
The anti-apoptotic survival functions of the cIAPs have been intensively researched, and their ability to either directly or indirectly inhibit caspase activation is well-described. From this standpoint, cIAP proteins are attractive targets for facilitating apoptosis in cancer cells. However, the presence of biallelic deletions, mutations and gene arrangements involving proteins within the TNFR1 complex have been associated with carcinogenesis in both multiple myelomaCitation43,Citation44 and splenic marginal zone lymphoma.Citation45 It has been hypothesized that cIAP inactivation supports tumor progression through NIK-mediated activation of IKKα and the alternative NFκB pathway.Citation43,Citation44 The activity of the alternative NFκB pathway is largely dependent on the steady-state level of NIK, which is constitutively targeted for degradation by a multi-subunit ubiquitin ligase complex composed of TNF receptor-associated factor 3 (TRAF3), TRAF2 and cIAP1/2.Citation6,Citation46,Citation47 Thus, mutations or alterations in any of these proteins could lead to accumulation of NIK, activation of IKKα and the alternative pathway.Citation6,Citation48-Citation50 Our results suggest that the cellular level of cIAPs are, in fact, precisely set to prevent stabilization of NIK. In MCF-10A (V12H-ras/cIAP2 shRNA) stable clones, in which only a partial reduction of cIAP2 was obtained after long-term culture, both NIK protein and NFκB activity were increased. Moreover, cIAP2 KD alone was sufficient to induce NFκB and inhibit p53. Consistent with this result, cIAP deletions in splenic marginal zone lymphomas are invariably monoallelic, indicative of the haploinsufficiency of these genes.Citation45
Although NIK was phosphorylated after cIAP2 KD, EMSA analysis of cIAP2 shRNA transfected cells demonstrated activation of the canonical pathway. Canonical pathway activation following treatment with IAP-antagonists has also been previously reported.Citation7 In IKKα/β−/− MEFs, MDM2 levels were shown to be regulated by IKKβ and canonical pathway signaling but not by IKKα.Citation24 Given this, cIAP2 KD and subsequent IKKα activation might not be expected to affect p53. It is likely that cIAP2 KD induced the appearance of canonical NFκB complexes as a result of IKKα-mediated IKKβ activation. Indeed, activated P-IKKα can mediate activation of IKKβ through transphosphorylation, resulting in strong activation of the heterodimeric kinase complex and the canonical pathway.Citation16,Citation51
MEFs lacking IKKα would represent similar conditions to our experiments involving KD of IKKα alone, which resulted in increased SUMO-MDM2. Thus, in IKKα−/− MEFs it would appear that IKKα does not have a role in regulation of MDM2 and p53. On the other hand, IKKβ would be indispensable in these cells for phosphorylation of MDM2 (Ser166) and induction of canonical NFκB. Importantly, IKKα KD alone increased SUMO-MDM2 and reduced SMP14-reactive MDM2 and p53 levels. This finding, combined with the ability of cIAP2 KD to activate IKKα, suggests that IKKα KD and activation of IKKα/-β have qualitatively similar effects on MDM2. Thus, IKKα can repress MDM2 SUMOylation in a non-catalytic manner. Interestingly, the MDM2 SUMO ligase PIAS1 can also be phosphorylated by activated IKKα on Ser90, which results in dissociation of a PIAS1:IKKα complex.Citation38 Immunoprecipitation of both PIAS1 and IKKα showed that these proteins interact in control transfected cells but not in cIAP2-depleted cells in which IKKα is activated. Therefore, either activation of IKKα or IKKα KD would eliminate the IKKα:PIAS1 interaction and potentially facilitate SUMOylation of MDM2. When both cIAP2 and PIAS1 were knocked-down, we observed a small decrease in SUMO-MDM2 accompanied by a substantial reduction in 95kDa P-MDM2 (Ser166) compared with cIAP2 siRNA alone. There are several possibilities that might account for these results. First, our results indicate that the 95 kDa P-MDM2 (Ser166) is less stable relative to SUMO-MDM2, since it was highly sensitive to transcriptional inhibition. Moreover, depending on the kinetics of each siRNA-mediated KD, it is possible that significant levels of P-MDM2 (Ser166) are SUMOylated and stabilized before the effect of PIAS1 KD occurs. Nonetheless, we cannot rule out that since multiple SUMO E3 ligases have been identified for MDM2,Citation22 PIAS1 KD alone may not be sufficient to prevent the SUMOylation of the entire pool of MDM2.
MCF10-AT1 cells have a constitutive level of NFκB activity, and our experiments suggest that IKKβ actively promotes the phosphorylation of MDM2 at Ser166 with or without cIAP2 KD in these cells. The underlying mechanism is not clear, but could involve IKKβ itself or a downstream kinase. Pim-2, a kinase capable of MDM2-Ser166 phosphorylation in response to growth factor signaling,Citation52 is regulated by NFκBCitation53 and could be a candidate kinase for NFκB-inducible MDM2-Ser166 phosphorylation.
V12H-ras-induced colony formation was enhanced after knockdown of cIAP2 in association with increased NFκB and decreased p53 protein. However, it is important to note that the interaction between p53 and IKKs as well as NFκB is complex and context-dependent. For example, the RelA and p52 subunits can directly interact with p53 to function as transcriptional co-regulators.Citation54 Key targets of this integrated transcriptional regulation include genes involved in control of cell death and proliferation, including SKP2 and p21WAF1 55 and p53AIP1.Citation56 In each case, the regulation of the gene by wtp53 was modified by the presence of NFκB, and co-regulation resulted in the augmentation of growth inhibitory and/or proapototic gene expression. NFκB transcriptional coactivation with wtp53 has most often been shown in cells in which p53 has been activated as part of the DNA damage respons.eCitation55,Citation57 DNA damage can result in post-translational modifications of NFκB subunits that augment the p53/NFKB2 (p52) collaborative response.Citation55 In contrast, NFκB has previously been shown to potentiate V12H-ras-mediated transformation of mouse fibroblasts through a mechanism involving co-regulation of ras-induced genes.Citation58 Moreover, similar to our results in non-transformed MCF-10A cells, constitutive NFκB activity delays V12H-ras-induced premature senescence.Citation59 Overall, the outcome of increased constitutive NFκB activity following cIAP2 knockdown, resulting in the downregulation of p53, would be reduction of cooperative growth inhibitory signaling between p53 and NFκB.
While our results show that the IKKs can serve to activate MDM2 and reduce p53 protein levels, competing mechanisms associated with oncogene expression also induce p53. Oncogene-mediated induction of p14ARF-/p19ARF inhibits MDM2 function. Moreover, oncogene-induction of stress-associated ribosome biogenesis can result in ribosomal protein binding to MDM2 resulting in its inactivation.Citation60,Citation61 Interestingly, the latter pathway appears to be specifically activated following oncogenic c-myc but not H-ras-mediated transformationCitation62 and therefore not a factor in the V12H-ras transformed MCF-10A cells in our study. Clearly, induction of the IKKs, in this instance resulting from knockdown of cIAP2, can override the MDM2 inactivating pathways to reduce p53 which would explain, at least in part, the NFκB contribution to cellular transformation.
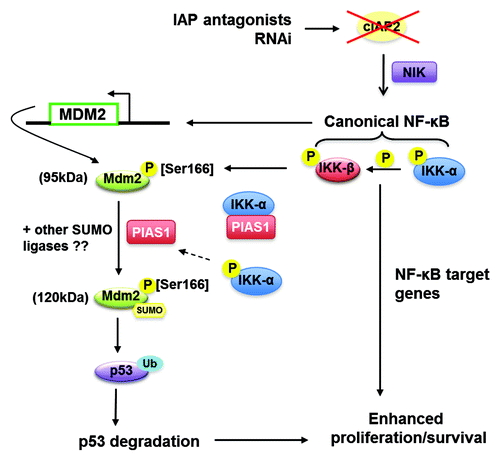
In summary, we present a model in that depicts the roles of IKKα and IKKβ following KD of cIAP2 in promoting the transcriptional activation and posttranslational modification of MDM2 to facilitate the ultimate degradation of p53. Our in vitro evidence shows that a reduction in cellular cIAP2 is sufficient to promote oncogene-induced colony formation in MCF-10AT1 cells that express wild-type p53. Consistent with this cIAP2 disruption has been found most often in high-risk chronic lymphocytic leukemia in which p53 is wild-type.Citation63 Thus, cIAP2 mutation or partial reduction could have ramifications on the promotion of various cancers resulting not only from IKKα-initiated NFκB activation, but also the downregulation of wild-type p53.
Figure 6. Hypothetical model of cIAP2-dependent regulation of p53. cIAP2 reduction results in the phosphorylation of IKKα which then activates IKKβ resulting in canonical NFκB activity. NFκB promotes a transient increase in MDM2 transcription, increasing the pool of MDM2 while IKKβ promotes MDM2 phosphorylation at Ser166. An IKKα:PIAS1 interaction is disrupted after cIAP2 KD and IKKα activation, which may contribute to the SUMOylation of MDM2. Induction and activation of MDM2 promotes the degradation of p53, which cooperates with NFκB to support cellular proliferation and survival.
Materials and Methods
Cells culture
MCF-10A human mammary epithelial cells were purchased from the American Type culture collection. MCF-10AT1 cells (derived from xenograft of V12H-ras transfected MCF-10A cellsCitation26) were obtained from Dr. L. Murphy, University of Manitoba. MCF-10A and MCF-10AT1 cells were maintained in Ham’s F12:DMEM (1:1) (GIBCO), 20 ng/mL epidermal growth factor (EGF) (Sigma), 10 μg/mL insulin (Sigma), 500 ng/mL hydrocortisone (Sigma) and 5% horse serum (GIBCO).
Transfections
Reverse transfections were performed for siRNA transfections. In 60 mm dishes, 1 mL of serum-free medium was mixed with 5 nM of the indicated siRNA and 5 μL of Dharmafect I (Dharmacon), then incubated at room temperature for 20 min. Cells were seeded on top of the transfection mixture. Cells were grown for 48 h prior to harvesting. The IκBαSR (the kind gift of Dr. A.S. Baldwin) plasmid was transfected into MCF-10AT1 cells using Fugene6 (Roche) and 18 h later, cells underwent reverse transfection with cIAP2 siRNA as described above.
siRNA sequences
siRNA targeted at cIAP1 (SMARTpool #M-004390–02–0005), cIAP2 (SMARTpool #M-004099–02–0005), XIAP (SMARTpool #M-004098–01–0005, IKKα #4 (siGENOME #D-003473–04–0005), IKKα #5 (siGENOME #D-003473–05–0005), PIAS1 (SMARTpool #M-008167–01–0005), non-targeting siRNA #1 (siGENOME #D-001210–01–05) were purchased from Dharmacon.
Colony-forming assay
MCF-10A cells were cotransfected using Fugene6 with oncogenic V12H-ras cDNA (gift of Dr. D. Gray, Ottawa Regional Cancer Centre) with either a control NT shRNA or a human cIAP2-shRNA construct. Cells were selected in 500 μg/ml G418 and colonies with a minimum of 50 cells were enumerated after staining with 0.2% crystal violet. Separate plates were used for colony isolation. Clones with reduced cIAP2 and overexpression of H-ras were expanded.
Treatments
MCF-10AT1 cells were treated with 10 μM Nutlin-3a (Sigma) for 3 h and/or 1 μg/mL Actinomycin D (Life Technologies) for 3 h prior to harvesting protein in RIPA buffer. For some experiments, 2 μM MG132 (Sigma) was added for the final 2 h of culture.
Adenovirus infection
Cells were plated in complete medium in 60 mm dishes. Twenty-four hours later, cells were infected with adenovirus expressing a kinase-dead mutant (K44A) of IKKβ64 at 25 MOI. Adenovirus expressing GFP was used as an adenoviral control. Protein was extracted 24 h later in RIPA buffer and analyzed by western blotting.
Immunoblots and antibodies
Whole-cell extracts were prepared in RIPA buffer and western blotting was performed as previously described.Citation65 Tumor samples were processed as whole tumor extracts for immunoblot analysis as previously described.Citation65 The following primary antibodies were used for western blot analysis: anti-actin #A-2066, and anti-Mdm2 SMP14 #M4308 and anti-FLAG (M2) were from Sigma; anti-cIAP1 #AF818 was from R&D Systems; anti-cIAP2 #S2700 was from Epitomics; anti-GADD45-α (C-4) #sc-6850, anti-phospho-NIK (Thr559) #sc-12957, anti-p21 (C-19) #sc-397, anti-p53 (BP-53) #sc-263 were from Santa Cruz; anti-IKKα #2682, anti-IKKβ #2684, phospho-IKKα/β (Ser176/180) #2697, anti-phospho-Mdm2 (Ser166) #3521, anti-NIK #4994, and anti-phospho-p53 (Ser15) #9284 were from Cell Signaling; anti-PIAS1 #ab32219 and anti-H-Ras #ab32417 were from Abcam; anti-rIAP1to detect cIAP1 and cIAP2, and anti-rIAP3 to detect XIAP were the kind gift of Dr. R. Korneluk and described.Citation66
To quantify the bands obtained via western blot analysis, ImageJ software based analysis (www.rsb.info.nih.gov/ij/) was used. The area under curve (AUC) of the specific signal was corrected for the AUC of the loading control. The data are representative of independent experiments performed at a minimum of three times.
q-RT-PCR
Cells were seeded in triplicate on 35 mm dishes. Total RNA extraction was performed using Trizol (Invitrogen) according to the manufacturer’s instructions. Reverse transcription-quantitative PCR (RT-qPCR) reactions were performed using the QuantiTect SYBR Green RT-PCR kit (Qiagen) with 100 ng of RNA and 0.5 uM of the respective primers, according to manufacturer's protocol. Forward and reverse primers for MDM2 and GAPDH were purchased from Invitrogen. Primer sequences for MDM2 were: forward 5′-GACCCTGGTTAGACCAAAGC-3′ and reverse-5′-CACGCCAAACAAATCTCCTA-3′and for GAPDH: forward-5′-CTCTCTGCTCCTCCTGTTCG-3′ and reverse-5′-ACGACCAAATCC GTTGACTC-3′. Reverse transcription was performed at 50°C for 30 min followed by initial PCR activation at 95°C for 15 min. Forty cycles consisting of a denaturing step at 94°C for 15 sec, an annealing step at 55°C for 30 sec, then an extension step at 72°C for 30 sec were performed.
Co-immunoprecipitation (co-IP)
MCF-10AT1 cells transfected with siRNA were harvested in co-IP buffer (25 mM Tris-Cl pH7.5, 150 mM NaCl, 50 mM NaF, 0.5 mM EDTA pH8, 0.5% Triton-X, 5 mM β-glycerophosphate, 5% glycerol, 1 mM DTT, 1 mM PMSF and 1 mM NaVO3). Three hundred μg of protein was incubated with 2 μg of the indicated antibody, or rabbit IgG as a control for 2 h at 4°C with rotation. Protein A/G agarose beads (Roche) were added and incubated overnight at 4°C with rotation. Agarose beads bound with immunoreactive complexes were washed four times with co-IP buffer. Following the final wash, immunoprecipitated complexes were eluted with 60 μL of 2x sample buffer and boiled for 5 min. Twenty-five μL of each sample was analyzed using SDS-PAGE as described above with 15 μg of input protein.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were isolated and subjected to EMSA using NFκB site oligonucleotides obtained from Promega as previously described.Citation65 Equivalence of extract loading was demonstrated by EMSA with a DNA fragment corresponding to the consensus Sp1 binding site (Promega). Supershift analysis was performed by incubating 2 μg of α-p50 (Millipore) with nuclear extract prior to addition of the probe.
Additional material
Download Zip (176.7 KB)Acknowledgments
The authors thank Drs. Herman Cheung and Michael McBurney for helpful discussion, Dr. Robert Korneluk for the gift of SM164. This work was supported by a grant from the Canadian Institutes of Health Research grant FRN 79304 (to M.A.C.P.)
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 2006; 7:988 - 94; http://dx.doi.org/10.1038/sj.embor.7400795; PMID: 17016456
- Choi YE, Butterworth M, Malladi S, Duckett CS, Cohen GM, Bratton SB. The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7 via unique mechanisms at distinct steps in their processing. J Biol Chem 2009; 284:12772 - 82; http://dx.doi.org/10.1074/jbc.M807550200; PMID: 19258326
- Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA 2008; 105:11778 - 83; http://dx.doi.org/10.1073/pnas.0711122105; PMID: 18697935
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30:689 - 700; http://dx.doi.org/10.1016/j.molcel.2008.05.014; PMID: 18570872
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131:669 - 81; http://dx.doi.org/10.1016/j.cell.2007.10.030; PMID: 18022362
- Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008; 9:1371 - 8; http://dx.doi.org/10.1038/ni.1676; PMID: 18997794
- Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007; 131:682 - 93; http://dx.doi.org/10.1016/j.cell.2007.10.037; PMID: 18022363
- Wright CW, Duckett CS. Reawakening the cellular death program in neoplasia through the therapeutic blockade of IAP function. J Clin Invest 2005; 115:2673 - 8; http://dx.doi.org/10.1172/JCI26251; PMID: 16200201
- Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125:1253 - 67; http://dx.doi.org/10.1016/j.cell.2006.05.030; PMID: 16814713
- Xu L, Zhu J, Hu X, Zhu H, Kim HT, LaBaer J, et al. c-IAP1 cooperates with Myc by acting as a ubiquitin ligase for Mad1. Mol Cell 2007; 28:914 - 22; http://dx.doi.org/10.1016/j.molcel.2007.10.027; PMID: 18082613
- Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res 2004; 64:7183 - 90; http://dx.doi.org/10.1158/0008-5472.CAN-04-1918; PMID: 15492230
- LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene 2008; 27:6252 - 75; http://dx.doi.org/10.1038/onc.2008.302; PMID: 18931692
- Fulda S. Targeting inhibitor of apoptosis proteins (IAPs) for cancer therapy. Anticancer Agents Med Chem 2008; 8:533 - 9; PMID: 18537535
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 2006; 441:431 - 6; http://dx.doi.org/10.1038/nature04870; PMID: 16724054
- Bassères DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene 2006; 25:6817 - 30; http://dx.doi.org/10.1038/sj.onc.1209942; PMID: 17072330
- Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE 2006; 2006:re13; http://dx.doi.org/10.1126/stke.3572006re13; PMID: 17047224
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25:280 - 8; http://dx.doi.org/10.1016/j.it.2004.03.008; PMID: 15145317
- Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 2001; 98:11598 - 603; http://dx.doi.org/10.1073/pnas.181181198; PMID: 11504915
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 2001; 3:973 - 82; http://dx.doi.org/10.1038/ncb1101-973; PMID: 11715018
- Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem 2002; 277:21843 - 50; http://dx.doi.org/10.1074/jbc.M109745200; PMID: 11923280
- Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003; 300:342 - 4; http://dx.doi.org/10.1126/science.1080386; PMID: 12690203
- Miyauchi Y, Yogosawa S, Honda R, Nishida T, Yasuda H. Sumoylation of Mdm2 by protein inhibitor of activated STAT (PIAS) and RanBP2 enzymes. J Biol Chem 2002; 277:50131 - 6; http://dx.doi.org/10.1074/jbc.M208319200; PMID: 12393906
- Lee MH, Lee SW, Lee EJ, Choi SJ, Chung SS, Lee JI, et al. SUMO-specific protease SUSP4 positively regulates p53 by promoting Mdm2 self-ubiquitination. Nat Cell Biol 2006; 8:1424 - 31; http://dx.doi.org/10.1038/ncb1512; PMID: 17086174
- Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell 2002; 1:493 - 503; http://dx.doi.org/10.1016/S1535-6108(02)00068-5; PMID: 12124178
- Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci USA 2009; 106:2629 - 34; http://dx.doi.org/10.1073/pnas.0812256106; PMID: 19196987
- Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol 1996; 148:313 - 9; PMID: 8546221
- Conze DB, Albert L, Ferrick DA, Goeddel DV, Yeh WC, Mak T, et al. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol 2005; 25:3348 - 56; http://dx.doi.org/10.1128/MCB.25.8.3348-3356.2005; PMID: 15798218
- Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol 2009; 1:a000950; http://dx.doi.org/10.1101/cshperspect.a000950; PMID: 20457558
- Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med 2010; 16:528 - 36; http://dx.doi.org/10.1016/j.molmed.2010.09.002; PMID: 20932800
- Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, et al. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res 2008; 68:9384 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-08-2655; PMID: 19010913
- Varfolomeev E, Alicke B, Elliott JM, Zobel K, West K, Wong H, et al. X chromosome-linked inhibitor of apoptosis regulates cell death induction by proapoptotic receptor agonists. J Biol Chem 2009; 284:34553 - 60; http://dx.doi.org/10.1074/jbc.M109.040139; PMID: 19854829
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 8; http://dx.doi.org/10.1126/science.1092472; PMID: 14704432
- de Toledo SM, Azzam EI, Dahlberg WK, Gooding TB, Little JB. ATM complexes with HDM2 and promotes its rapid phosphorylation in a p53-independent manner in normal and tumor human cells exposed to ionizing radiation. Oncogene 2000; 19:6185 - 93; http://dx.doi.org/10.1038/sj.onc.1204020; PMID: 11175332
- Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10:501 - 14; http://dx.doi.org/10.1016/j.ccr.2006.10.010; PMID: 17157790
- Xirodimas DP, Chisholm J, Desterro JM, Lane DP, Hay RT. P14ARF promotes accumulation of SUMO-1 conjugated (H)Mdm2. FEBS Lett 2002; 528:207 - 11; http://dx.doi.org/10.1016/S0014-5793(02)03310-0; PMID: 12297306
- Meek DW, Knippschild U. Posttranslational modification of MDM2. Mol Cancer Res 2003; 1:1017 - 26; PMID: 14707285
- Schmidt D, Müller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci USA 2002; 99:2872 - 7; http://dx.doi.org/10.1073/pnas.052559499; PMID: 11867732
- Liu B, Yang Y, Chernishof V, Loo RR, Jang H, Tahk S, et al. Proinflammatory stimuli induce IKKalpha-mediated phosphorylation of PIAS1 to restrict inflammation and immunity. Cell 2007; 129:903 - 14; http://dx.doi.org/10.1016/j.cell.2007.03.056; PMID: 17540171
- Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 2002; 22:3497 - 508; http://dx.doi.org/10.1128/MCB.22.10.3497-3508.2002; PMID: 11971980
- Liu Z, Li H, Derouet M, Filmus J, LaCasse EC, Korneluk RG, et al. ras Oncogene triggers up-regulation of cIAP2 and XIAP in intestinal epithelial cells: epidermal growth factor receptor-dependent and -independent mechanisms of ras-induced transformation. J Biol Chem 2005; 280:37383 - 92; http://dx.doi.org/10.1074/jbc.M503724200; PMID: 16115895
- Ma O, Cai W-W, Zender L, Dayaram T, Shen J, Herron AJ, et al. MMP13, Birc2 (cIAP1), and Birc3 (cIAP2), amplified on chromosome 9, collaborate with p53 deficiency in mouse osteosarcoma progression. Cancer Res 2009; 69:2559 - 67; http://dx.doi.org/10.1158/0008-5472.CAN-08-2929; PMID: 19276372
- Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, et al. NF-kappa B-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proc Natl Acad Sci USA 2004; 101:13618 - 23; http://dx.doi.org/10.1073/pnas.0402069101; PMID: 15353598
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007; 12:131 - 44; http://dx.doi.org/10.1016/j.ccr.2007.07.003; PMID: 17692805
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007; 12:115 - 30; http://dx.doi.org/10.1016/j.ccr.2007.07.004; PMID: 17692804
- Rossi D, Deaglio S, Dominguez-Sola D, Rasi S, Vaisitti T, Agostinelli C, et al. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood 2011; 118:4930 - 4; http://dx.doi.org/10.1182/blood-2011-06-359166; PMID: 21881048
- Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 2004; 279:26243 - 50; http://dx.doi.org/10.1074/jbc.M403286200; PMID: 15084608
- Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol 2008; 9:1364 - 70; http://dx.doi.org/10.1038/ni.1678; PMID: 18997792
- He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, et al. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med 2006; 203:2413 - 8; http://dx.doi.org/10.1084/jem.20061166; PMID: 17015635
- Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity 2004; 21:629 - 42; http://dx.doi.org/10.1016/j.immuni.2004.09.011; PMID: 15539150
- Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood 2010; 115:3541 - 52; http://dx.doi.org/10.1182/blood-2009-09-243535; PMID: 20053756
- O’Mahony A, Lin X, Geleziunas R, Greene WC. Activation of the heterodimeric IkappaB kinase alpha (IKKalpha)-IKKbeta complex is directional: IKKalpha regulates IKKbeta under both basal and stimulated conditions. Mol Cell Biol 2000; 20:1170 - 8; http://dx.doi.org/10.1128/MCB.20.4.1170-1178.2000; PMID: 10648602
- Meek DW, Hupp TR. The regulation of MDM2 by multisite phosphorylation--opportunities for molecular-based intervention to target tumours?. Semin Cancer Biol 2010; 20:19 - 28; http://dx.doi.org/10.1016/j.semcancer.2009.10.005; PMID: 19897041
- Li J, Peet GW, Balzarano D, Li X, Massa P, Barton RW, et al. Novel NEMO/IkappaB kinase and NF-kappa B target genes at the pre-B to immature B cell transition. J Biol Chem 2001; 276:18579 - 90; http://dx.doi.org/10.1074/jbc.M100846200; PMID: 11279141
- Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer 2012; 12:121 - 32; PMID: 22257950
- Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NF-kappaB subunit. EMBO J 2006; 25:4820 - 32; http://dx.doi.org/10.1038/sj.emboj.7601343; PMID: 16990795
- O’Prey J, Crighton D, Martin AG, Vousden KH, Fearnhead HO, Ryan KM. p53-mediated induction of Noxa and p53AIP1 requires NFkappaB. Cell Cycle 2010; 9:947 - 52; http://dx.doi.org/10.4161/cc.9.5.10872; PMID: 20160496
- Oren M, Cooks TNF. NFκB and p53: A life and death affair. Cell Cycle 2010; 9:1027; http://dx.doi.org/10.4161/cc.9.6.11099; PMID: 20237416
- Azmi AS, Banerjee S, Ali S, Wang Z, Bao B, Beck FW, et al. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget 2011; 2:378 - 92; PMID: 21623005
- Hanson JL, Hawke NA, Kashatus D, Baldwin AS. The nuclear factor kappaB subunits RelA/p65 and c-Rel potentiate but are not required for Ras-induced cellular transformation. Cancer Res 2004; 64:7248 - 55; http://dx.doi.org/10.1158/0008-5472.CAN-03-3898; PMID: 15492243
- Batsi C, Markopoulou S, Vartholomatos G, Georgiou I, Kanavaros P, Gorgoulis VG, et al. Chronic NF-kappaB activation delays RasV12-induced premature senescence of human fibroblasts by suppressing the DNA damage checkpoint response. Mech Ageing Dev 2009; 130:409 - 19; http://dx.doi.org/10.1016/j.mad.2009.04.002; PMID: 19406145
- Miliani de Marval PL, Zhang Y. The RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget 2011; 2:234 - 8; PMID: 21406728
- Pan W, Issaq S, Zhang Y. The in vivo role of the RP-Mdm2-p53 pathway in signaling oncogenic stress induced by pRb inactivation and Ras overexpression. PLoS One 2011; 6:e21625; http://dx.doi.org/10.1371/journal.pone.0021625; PMID: 21747916
- Rossi D, Fangazio M, Rasi S, Vaisitti T, Monti S, Cresta S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood 2012; 119:2854 - 62; http://dx.doi.org/10.1182/blood-2011-12-395673; PMID: 22308293
- Sanlioglu S, Williams CM, Samavati L, Butler NS, Wang G, McCray PB Jr., et al. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-α secretion through IKK regulation of NF-κ B. J Biol Chem 2001; 276:30188 - 98; http://dx.doi.org/10.1074/jbc.M102061200; PMID: 11402028
- Pratt MA, Bishop TE, White D, Yasvinski G, Ménard M, Niu MY, et al. Estrogen withdrawal-induced NF-kappaB activity and bcl-3 expression in breast cancer cells: roles in growth and hormone independence. Mol Cell Biol 2003; 23:6887 - 900; http://dx.doi.org/10.1128/MCB.23.19.6887-6900.2003; PMID: 12972607
- Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 1996; 379:349 - 53; http://dx.doi.org/10.1038/379349a0; PMID: 8552191