Abstract
The rate of inherent resistance to single-agent trastuzumab in HER2-overexpressing metastatic breast carcinomas is impressive at above 70%. Unfortunately, little is known regarding the distinctive genetic signatures that could predict trastuzumab refractoriness ab initio. The epithelial-to-mesenchymal transition (EMT) molecular features, HER2 expression status and primary responses to trastuzumab were explored in the public Lawrence Berkeley Laboratory (LBL) Breast Cancer Collection. Lentivirus-delivered small hairpin RNAs were employed to reduce specifically and stably the expression of EMT transcription factors in trastuzumab-refractory basal/HER2+ cells. Cell proliferation assays and pre-clinical nude mice xenograft-based studies were performed to assess the contribution of specific EMT transcription factors to inherent trastuzumab resistance. Primary sensitivity to trastuzumab was restricted to the SLUG/SNAIL2-negative subset of luminal/HER2+ cell lines, whereas all of the SLUG/SNAIL2-positive basal/HER2+ cell lines exhibited an inherent resistance to trastuzumab. The specific knockdown of SLUG/SNAIL2 suppressed the stem-related CD44+CD24-/low mesenchymal immunophenotype by transcriptionally upregulating the luminal epithelial marker CD24 in basal/HER2+ cells. Basal/HER2+ cells gained sensitivity to the growth-inhibitory effects of trastuzumab following SLUG/SNAIL2 gene depletion, which induced the expression of the mesenchymal-to-epithelial transition (MET) genes involved in promoting an epithelial phenotype. The isolation of CD44+CD24-/low mesenchymal cells by magnetic-activated cell sorting (MACS) confirmed their intrinsic unresponsiveness to trastuzumab. A reduction in tumor growth and dramatic gain in sensitivity to trastuzumab in vivo were confirmed when the SLUG/SNAIL2 knockdown basal/HER2+ cells were injected into nude mice. HER2 overexpression in a basal, rather than in a luminal molecular background, results in a basal/HER2+ breast cancer subtype that is intrinsically resistant to trastuzumab. EMT transcription factors might induce an enhanced phenotypic plasticity that would allow basal/HER2+ breast cancer cells to “enter” into and “exit” dynamically from trastuzumab-responsive stem cell-like states. The systematic determination of SLUG/SNAIL2 as a stem/CD44+CD24-/low cell-associated protein may improve the therapeutic management of HER2+ breast carcinomas.
Introduction
The HER2 gene plays a crucial role in the natural history of human breast cancer due to its unique ability to drive the pool of replenishing cancer-initiating or -propagating cells, which have the unique ability to both self-renew and give rise to phenotypically heterogeneous breast cancer progeny and are popularly known as cancer stem cells (CSCs).Citation1-Citation5 The CS-like cells isolated from breast carcinomas intrinsically possess a high resistance to hormonal therapy, chemotherapy and radiotherapy and, consequently, appear to be highly prevalent at the invasive edge of breast carcinomas following treatment with conventional therapies.Citation6-Citation11 However, the effects of HER2 overexpression on breast CSCs (e.g., the upregulation of the stem cell marker aldehyde dehydrogenase, increased expression of stem cell regulatory genes and increased tumorigenesis in severely immunocompromised mice) can be blocked by the anti-HER2 monoclonal antibody trastuzumab in sensitive but not resistant breast cancer cell lines.Citation1,Citation12,Citation13 Similarly, the EGFR/HER2 dual tyrosine kinase inhibitor lapatinib has also been shown to decrease the subpopulation of chemotherapy-resistant cancer-initiating cells in breast carcinomas in vivo.Citation6 Therefore, the clinical efficacy of trastuzumab may largely relate to its ability to target the CSC population in HER2-amplified tumors specifically and efficiently.Citation1,Citation6,Citation14,Citation15 Moreover, Magnifico et al.Citation16 further expanded this notion to demonstrate that breast CSCs are exquisitely sensitive to trastuzumab, because they appear to express the highest levels of the HER2 protein, regardless of their HER2 gene amplification status.
The crucial finding that HER2 may be selectively upregulated in breast CSCs but not in bulk breast cancer cell populations might explain why trastuzumab may be effective when administered in the adjuvant setting of breast tumors classified as HER2-negative [i.e., immunohistochemistry (IHC) 1/2+ or fluorescence in situ hybridization (FISH) negative].Citation17,Citation18 In breast tumors clinically classified as bona fide HER2-positive (i.e., IHC 3+ or FISH-positive), HER2 is overexpressed in both breast CSCs and bulk tumor populations; therefore, they are expected to be sensitive to HER2-targeting agents, which are expected to induce tumor regression and reduce recurrence in advanced and adjuvant settings, respectively.Citation18 Although the addition of trastuzumab to adjuvant chemotherapy can impressively reduce the recurrence rate by almost 50% and improve survival in breast cancer patients with early-stage HER2-positive tumors, approximately 15% of women diagnosed with early HER2-positive disease exhibit de novo (i.e., primary or intrinsic) resistance to trastuzumab and relapse in spite of the treatment with trastuzumab-based therapies.Citation19-Citation25 Moreover, the overall response rate of women diagnosed with HER2-positive metastatic breast cancer and treated with trastuzumab as a single first-line agent is only 26%; that is, > 70% of HER2-overexpressing metastatic breast carcinomas show a resistance to trastuzumab ab initio. A variety of possible mechanisms of escape from trastuzumab appear to involve many of the same biomarkers that have been implicated in the biology of CS-like cells: e.g., the overexpression of the stem cell-related marker CD44, leading to a loss or blockage of the trastuzumab-binding site at the extracellular domain of HER2;Citation26,Citation27 the upregulation of stem cell markers, such as CXCR4, β1 integrin or Notch-1,Citation28-Citation32 leading to the activation of alternative pathways circumventing HER2 signaling and the upregulation of pro-survival mediators, such as the inhibitor of apoptosis survivin.Citation33 Accordingly, it has been suggested that, although trastuzumab effectively targets cancer-initiating cells, a clinical resistance to trastuzumab may counter-intuitively be driven by breast CSCs.Citation34
We have recently hypothesized that, when HER2 gene amplification, generally within differentiated luminal breast cancer phenotypes, occurs in a basal molecular background, it results in a basal/HER2+ subtype of breast carcinomas that naturally exhibit an inherent (i.e., primary) resistance to trastuzumab.Citation35 Mechanistically, an intrinsic tumor cell plasticity able to efficiently drive the emergence of a CS-related CD44+CD24-/low mesenchymal phenotype might account for the de novo resistance to trastuzumab in basal/HER2+ breast carcinomas.Citation12,Citation36 By stably knocking down the expression of several epithelial-to-mesenchymal transition (EMT) transcription factors in de novo trastuzumab-resistant HER2+ breast cancer cells, we suggest, for the first time, that an intrinsic phenotypic plasticity in basal/HER2+ breast cancer cells may permit them to “enter” into and “exit” dynamically from trastuzumab-sensitive stem cell-like states.
Results
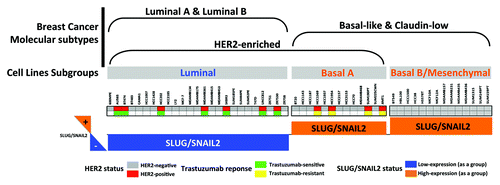
Overexpression of the EMT regulator SLUG/SNAIL2 is coincidental with a “basal/HER2+” phenotype in breast cancer cells with primary resistance to trastuzumab
We took advantage of previous studies that aimed to summarize the molecular and cellular characteristics of EMT in the entire set of breast cancer cell lines originally included in the “Neve data.”Citation37,Citation38 When we examined the expression status of the EMT transcriptional driver SLUG/SNAI2 in the 51 breast cancer cell lines organized by subclasses, as defined in Neve et al.Citation39 (i.e., luminal, basal A and basal B), most of the HER2 gene-amplified breast carcinomas cell lines (i.e., AU565, BT474, HCC202, MDA-MB-361, SKBR3, UACC812 and ZR7530) were found to belong to the SLUG/SNAIL2-negative luminal subclass of breast tumors (). Although the entire subset of mesenchymal-like basal B cell lines also lacked the amplification of the HER2 gene, a few HER2 gene-amplified breast cancer cell lines matched both the luminal subgroup and the basal A subgroup of cell lines (i.e., HCC1569, HCC1954 and SUM190T). Of note, when the HER2-positive breast cancer cell lines were classified as “trastuzumab-sensitive” or “trastuzumab-refractory” based on the data from the literature, we observed that the trastuzumab sensitivity ab initio was restricted to the SLUG/SNAIL2-negative subset of luminal/HER2+ cell lines, whereas all of the SLUG/SNAIL2-positive basal/HER2+ cell lines exhibited a primary (inherent) resistance to trastuzumab (). Moreover, we examined JIMT-1 cells, which were not originally included in the Neve data set; these cells are derived from a ductal carcinoma pleural metastasis of a 62-year-old patient who did not respond to trastuzumab treatment despite having a tumor with a HER2 gene amplification.Citation40-Citation42 We found that the JIMT-1 cells also belonged to the SLUG/SNAIL2-positive subgroup of trastuzumab-refractory basal/HER2 breast cancer cell lines. However, the expression status of other EMT drivers (e.g., ZEB1) appeared to lack a discriminating role between trastuzumab responders and non-responders in luminal/HER2 and basal/HER2 breast cancer cell lines (data not shown).
Figure 1. Association of the clinical subtypes of breast cancer cell lines with the primary response to the anti-HER2 monoclonal antibody trastuzumab. The relation between the morphological subgroups of breast cancer cell lines (luminal, basal-A, basal-B/mesenchymal), and breast tumor subtypes (luminal A, luminal B, HER2-enriched, basal-like, claudin-low) was evaluated by assessing the status of HER2 gene amplification, the primary response to trastuzumab and the expression status of SLUG/SNAIL2 as discriminators. As a group, the trastuzumab-resistant basal/HER2+ cell lines overexpressed the EMT transcriptional factor SLUG/SNAIL2, whereas trastuzumab-sensitive luminal/HER2+ cell lines underexpressed SLUG/SNAIL2.
Trastuzumab-refractory basal/HER2 JIMT1 breast cancer cells are enriched for EMT features, including the transcriptional repressor SLUG/SNAIL2
We sought to confirm experimentally that the trastuzumab-refractory basal/HER2 JIMT1 cells were enriched for EMT features, including putative “trastuzumab response discriminators,” such as SLUG/SNAIL2. Using RT-PCR, we compared the expression status of four EMT markers [i.e., vimentin (VIM), E-cadherin, N-cadherin and fibronectin] and six EMT drivers (i.e., SNAIL1, SLUG/SNAIL2, SNAIL3, TWIST1, ZEB1 and ZEB2) in the trastuzumab-refractory basal/HER2 JIMT1 cells to the luminal/HER2-positive SKBR3, basal-like/HER2-negative MDA-MB-468 and post-EMT (mesenchymal)/HER2-negative MDA-MB-231 breast cancer cells ().
Figure 2. Relative enrichment of EMT features in trastuzumab-resistant basal/HER2+ JIMT1 breast cancer cells. Figure shows the relative expression of EMT-associated genes in basal/HER2+ JIMT1, basal A MDA-MB-468 and basal B MDA-MB-231 breast cancer cell vs. luminal/HER2+ SKBR3 breast cancer cells using the human epithelial-to-mesenchymal (EMT) RT2 Profiler PCR Array (PAHS-090, 96-well format) as per the manufacturer’s instructions (SABiosciences).
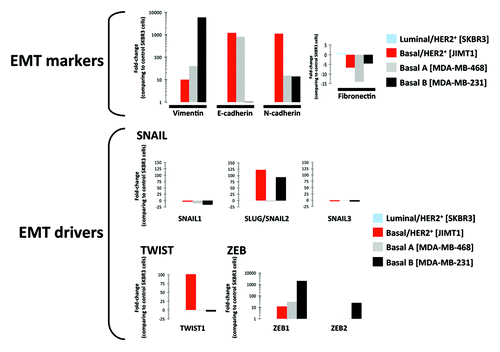
Although VIM expression is not proof of EMT, it was noteworthy that, despite maintaining an E-cadherin-positive epithelial phenotype, the trastuzumab-resistant basal/HER2+ JIMT1 cells expressed relatively high levels of VIM compared with the trastuzumab-sensitive SKBR3 cells (), which exhibited an E-cadherin negativity despite being luminal/HER2+ breast carcinoma cells due to a homozygous deletion at 16q22.1 that includes the CDH1 gene.Citation42 N-cadherin and fibronectin (FN), which are classically used to monitor EMT, showed relative enrichment and underexpression, respectively, in the basal/HER2 JIMT1 cells. The SNAIL family members SNAIL1 and SNAIL3 exhibited little difference among the SKBR3, MDA-MB-468 and MDA-MB-231 breast cancer cells. We similarly confirmed a more than 120-fold increase in the relative expression of SLUG/SNAIL2 in the trastuzumab-resistant basal/HER2+ JIMT-1 cells compared with the trastuzumab-responsive luminal/HER2+ SKBR3 cells. Of note, the relative upregulation of SLUG/SNAIL2 expression in the HER2 gene-amplified JIMT1 cells was comparable to the HER2-negative MDA-MB-231 mesenchymal cells, whereas the HER2-negative MDA-MB-468 basal-like cells did not show an increase in the expression of SLUG/SNAIL2. In a similar manner and in accordance with the ability of SLUG/SNAIL2 and TWIST1 to suppress the epithelial branch of the EMT program redundantly in a cooperative manner,Citation43 the relative expression of TWIST1 was increased by approximately 100-fold in the trastuzumab-resistant HER2+SLUG+ JIMT-1 cells compared with the trastuzumab-responsive HER2+SLUG- SKBR3 cells. HER2-negative MDA-MB-468 basal cells showed no increase in the expression of TWIST. The HER2+SLUG+ JIMT-1 cells and HER2-SLUG- MDA-MB-468 exhibited relatively high levels of ZEB1, the EMT transcription factor that was upregulated with the highest discriminator score in basal B/mesenchymal cells.
Knockdown of SLUG/SNAIL2 suppresses the CD44+CD24-/low phenotype in trastuzumab-refractory JIMT1 cells
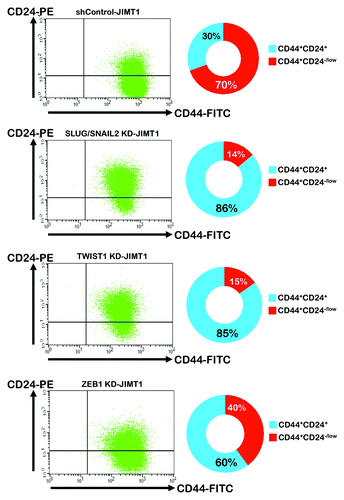
To evaluate the role of EMT drivers in regulating the CD44+CD24-/low mesenchymal subpopulationCitation44-Citation46 in basal/HER2+ JIMT1 cells, we used flow cytometry to determine the amount of JIMT1 cells bearing the CD44+CD24-/low mesenchymal immunophenotype before and after the lentiviral-mediated small hairpin (sh) RNA knockdowns of the EMT-driving transcription factors SLUG/SNAI2, TWIST1 and ZEB1. shows a summarized analysis of the shRNA control-JIMT1 cells and EMT knocked-down JIMT1 derivatives with respect to four fractions of cell subpopulations, as defined by the absence or presence of the CD44 and CD24 cell-surface markers. Of note, the knockdown of SLUG/SNAIL2 expression markedly reduced the amount of cells displaying the CD44+CD24-/low mesenchymal phenotype from approximately 70% in the parental JIMT1 cells to 14% (5-fold decrease) in the SLUG/SNAIL2 KD-JIMT1 cells, and virtually all of the resting cells displayed a CD44+CD24+ basal-epithelial phenotype (86%). The specific knockdown of TWIST1 notably mimicked the ability of the SLUG/SNAI2 knockdown to decrease the percentage of CD44+CD24-/low mesenchymal cells by approximately 5-fold. The specific knockdown of ZEB1 reduced the percentage of the JIMT1 cells bringing the CD44+CD24-/low mesenchymal immunophenotype from 70% to 40%, which was lower than the specific silencing of SLUG/SNAIL2 or TWIST1.
Figure 3. Impact of shRNA-driven genetic ablation of EMT transcription factors on CD44+CD24-/low cell surface markers in basal/HER2+ JIMT1 cells. Left parts: Provided are representative flow cytometry dot plots of CD44 and CD24 expression of JIMT1 cells engineered to stably exhibit knocked-down expression of SLUG/SNAIL2, TWIST1 or ZEB1. Right parts: The average percentage of CD44+CD24-/low (red) and CD44+CD24+ (blue) cells determined by flow cytometry was calculated from three independent experiments.
SLUG/SNAIL2 transcriptionally regulates the expression status of the luminal epithelial marker CD24
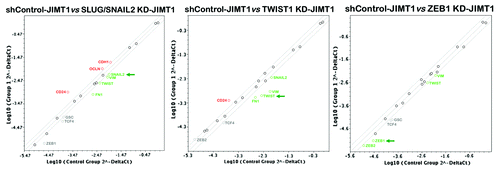
The expression of the luminal epithelial marker CD24 is known to be dynamically regulated:Citation44,Citation46,Citation47 CD44+CD24+ cells can readily give rise to CD44+CD24-/low cells and vice versa.Citation46 Thus, we sought to assess whether the repression of the CD44+CD24-/low mesenchymal phenotype that occurred in response to the specific silencing of the EMT driver SLUG/SNAIL2 was transcriptionally regulated in parallel to other genetic events controlling the EMT phenomenon, which can be defined by the rapid transcriptional repression of epithelial markers and the sustained induction of EMT-promoting transcription factors. The total RNAs from the shRNA control-JIMT1, SLUG/SNAIL2 KD-JIMT1, TWIST1 KD-JIMT1 and ZEB1 KD-JIMT1 cells were evaluated using a customized array for the quantitative real-time PCR (qRT-PCR) analysis of 19 genes specifically associated with EMT and the reciprocal mesenchymal-to-epithelial transition (MET) (see the Materials and Methods section for the genes included in the array; ). When a 2-fold or greater difference in the mRNA expression level was used as the cut-off to determine significant regulatory effects on the genes involved in the EMT genetic program, the SLUG/SNAIL2 KD-JIMT1 cells showed decreased mRNA levels of SLUG/SNAIL2, a result that was accompanied by the concomitant repression of the EMT driver TWIST1 and the EMT markers VIM and FN. Because SLUG/SNAIL2 is a member of the zinc-finger transcription factor family that binds to the E-cadherin (CDH1) gene promoter region to repress CDH1 transcription during EMT,Citation48,Citation49 it was not surprising to find that the SLUG/SNAIL2 knockdown correlated with the significantly enhanced transcription of the epithelial-specific gene markers CDH1 and OCLN [occludin or zonula occludens-1 (ZO-1)]. Of note, the CD24 mRNA expression was significantly and concomitantly upregulated, whereas the expression of the CD44 transcript remained unaltered in response to the SLUG/SNAIL2 knockdown. The stable knockdown of TWIST1 largely recapitulated the transcriptional effects observed upon the stable knockdown of SLUG/SNAIL2. The TWIST1 KD-JIMT1 cells were similarly found to have decreased mRNA levels of TWIST1, which was accompanied by the concomitant repression of the EMT driver SLUG/SNAIL2 and the EMT markers VIM and FN. The CD24 transcript level was also significantly increased in the TWIST1 KD-JIMT1 cells: the expression status of CD44 remained unaltered compared with the shRNA-control JIMT1 cells. The stable knockdown of ZEB1 was found to repress the expression of both ZEB1 and ZEB2 transcripts significantly, a result that was accompanied by the slight downregulation of TWIST1 and VIM. However, we failed to detect a significant increase in the expression of CD24 in the ZEB1 KD-JIMT1 cells.
Figure 4. Impact of shRNA-driven genetic ablation of EMT transcription factors on the epithelial-to-mesenchymal (EMT) genetic program in basal/HER2+ JIMT1 cells. Total RNA from shControl-JIMT1, SLUG/SNAIL2 KD-JIMT1, TWIST1 KD-JIMT1, ZEB1 KD-JIMT1 cells was characterized in technical triplicates using a customized PCR array as described in the Materials and Methods section. Figures show representative scatter plots of the difference (≥ 2-fold; green and red symbols indicate downregulation and upregulation, respectively vs. basal expression levels in shControl-transduced JIMT1 cells) in relative transcript abundance of VIM, E-cadherin, N-cadherin, fibronectin, SNAIL1, SLUG/SNAIL2, SNAIL3, TWIST1, ZEB1 and ZEB2. Grey symbols denote fold-change results that need to be validated with a sufficient number of biological replicates—i.e., fold-change results may have greater variations if p value > 0.05, or the p value for the fold-change is either unavailable or relatively high (p > 0.05)—or they are interpretable because gene’s average threshold cycles was either not determined or greater than the defined cut-off value (default 35) in both samples. KD, knockdown
Knockdown of SLUG/SNAIL2 sensitizes JIMT1 cells to the growth-inhibitory effects of trastuzumab
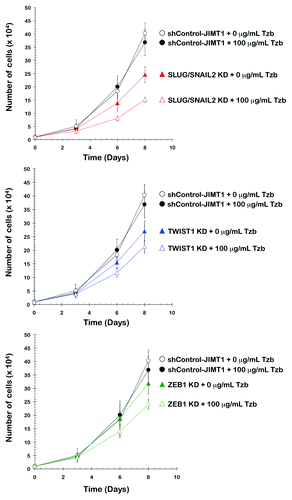
We measured cell proliferation by counting the cell numbers to determine whether the JIMT1 cell populations in which SLUG/SNAIL2 was stably knocked-down and that lacked the CD44+CD24-/low mesenchymal subpopulation exhibited an increased response to trastuzumab (). After 0, 3, 6 and 8 d of culture in the absence or presence of 100 μg/mL trastuzumab, the cells were collected and counted to investigate the effect of SLUG/SNAIL2 KD and trastuzumab on breast cancer cell numbers. The knockdown of the EMT driver SLUG/SNAIL2 was sufficient to reduce the cell numbers of the JIMT1 parental cells significantly. Moreover, supra-additive (synergistic) growth-inhibitory interactions seemed to occur at late time points (i.e., 6 and 8 d) when the SLUG/SNAIL2-knockdown cells were cultured in the presence of 100 μg/mL trastuzumab, an ineffective dose when used as a single agent in the parental JIMT1 cells (data not shown) and shRNA-control JIMT1 cells. Although less markedly than the SLUG/SNAIL2 KD-JIMT1 cells, the knockdown of TWIST1 significantly reduced the cell numbers of the JIMT1 cells in the presence of trastuzumab. The knockdown of ZEB1 was the least efficient EMT gene knockdown strategy for enhancing the anti-proliferative activity of trastuzumab against the trastuzumab-refractory JIMT1 cells.
Figure 5. Impact of the shRNA-driven genetic ablation of EMT transcription factors on trastuzumab-induced inhibition of breast cancer cell proliferation. shControl-JIMT1, SLUG/SNAIL2 KD-JIMT1, TWIST1 KD-JIMT1 and ZEB1 KD-JIMT1 cells were plated in 24-well plates at a density of 10,000 cells/well and cultured with regular media in the absence or presence of 100 μg/mL trastuzumab. The data presented are the means of number cells × 104/well and 95% confidence intervals (bars) from three independent experiments made in duplicate and obtained after 0, 3, 6 and 8 d. KD, knockdown; Tzb, trastuzumab.
The CD44+CD24-/low mesenchymal subpopulation is responsible for trastuzumab refractoriness in basal/HER2+ JIMT1 cells
To elucidate whether the sensitizing effects of the SLUG/SNAIL2 knockdown on the trastuzumab activity were due to a loss of the CD44+CD24-/low mesenchymal subpopulation, we examined whether the non-CD44+CD24-/low epithelial and CD44+CD24-/low mesenchymal cell subpopulations directly isolated from the trastuzumab-refractory basal/HER2 JIMT1 cell line displayed differential sensitivities to the growth-inhibitory effects of trastuzumab. First, the non-CD44+CD24-/low (CD44+CD24+-enriched; approx. 80%) and CD44+CD24-/low (> 95%) immunophenotypes were isolated using magnetic-activated cell sorting (MACS; ). The immunofluorescence analyses of the CD44+CD24-/low-sorted cells visually confirmed the overexpression of CD44 and no expression or weak expression of CD24. The immunofluorescence analysis also suggested an increase in VIM expression within the CD44+CD24-/low-sorted mesenchymal subpopulation, whereas the CD44+CD24+-enriched basal-epithelial cells mostly lacked VIM expression (). Unsorted, CD44+CD24+/low-depleted and CD44+CD24-/low-sorted JIMT1 cells were treated with graded concentrations of trastuzumab on the day after sorting. Interestingly, each cell subpopulation differed in its cytostatic responses to trastuzumab. Although the unsorted control cells and CD44+CD24-/low mesenchymal subpopulation largely failed to exhibit any significant response to trastuzumab, the non-CD44+CD24-/low cells gained some sensitivity to the cytostatic effects of trastuzumab, as their MTT uptake was significantly reduced by more than 50% in the presence of 100 μg/mL trastuzumab (, top). Of note, when the response to trastuzumab was evaluated in the CD44+CD24-/low mesenchymal subpopulations sorted from the SLUG/SNAIL2 KD-JIMT1 cells, it was found that these cells significantly lost their intrinsic resistance to trastuzumab (, top).
Figure 6. Immunohistochemical characterization of CD44+CD24-/low mesenchymal cells sorted from trastuzumab-refractory, basal/HER2+ JIMT1 cells. Trastuzumab-refractory, basal/HER2+ JIMT1 cells were sorted into CD44+CD24-/low-depleted and CD44+CD24-/low-enriched populations (top panels) and then stained for CD44, CD24 and the EMT marker VIM immediately following sorting (bottom panels). Enrichment of target cells by magnetic MicroBeads (MACS® Technology) was performed according to the manufacturer’s protocol. Representative immunofluorescent stainings of CD44 (red), CD24 (green), VIM (green) and Hoechst 33258 (blue) are presented. CD44+CD24-/low mesenchymal cells were found to have a high nucleo/cytoplasmic (N/C) ratio and a greater expression of VIM.
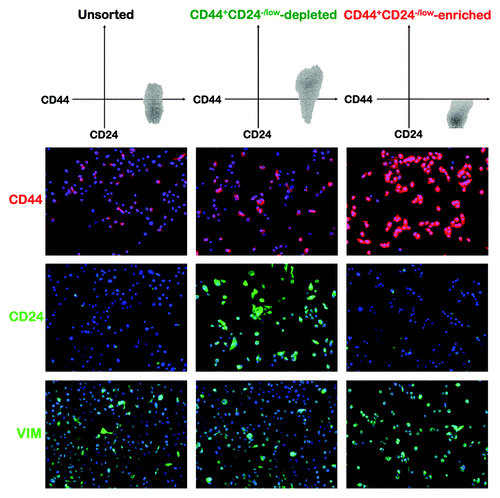
Figure 7. Impact of the shRNA-driven genetic ablation of SLUG/SNAIL2 on the efficacy of trastuzumab in vitro and in vivo. Top panels: Changes in the cell viability of trastuzumab-resistant JIMT1 cell populations depleted or enriched for CD44+CD24-/low mesenchymal cells. The metabolic status of unsorted JIMT1 parental cells, CD44+CD24-/low-depleted, and CD44+CD24-/low-enriched JIMT1 cells treated with graded concentrations of trastuzumab before and after SLUG/SNAIL2 knockdown was evaluated using MTT-based cell viability assays and constructing dose-response graphs as % of untreated cells (untreated control cells = 100% cell viability). Results are means (columns) and 95% confidence intervals (bars) of three independent experiments made in triplicate. Bottom panels: Changes in the tumor volume of shControl-JIMT1 and SLUG/SNAIL2 KD-JIMT1 xenografts grown in immunodeficient mice treated with trastuzumab (5 mg/kg/week).
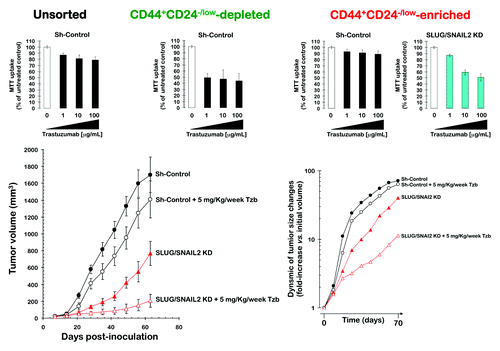
Knockdown of SLUG/SNAIL2 reduces tumor growth and sensitizes trastuzumab-resistant xenografts to trastuzumab
The effects of the SLUG/SNAIL2 knockdown on tumor growth were studied in vivo using a JIMT1 xenograft animal model (, bottom). Compared with the untreated control group (1,701 ± 210 mm3), nine weeks of treatment with trastuzumab (5 mg/kg/wk) failed to induce significant reductions in the mean tumor size (1,409 ± 220 mm3). However, compared with the mean xenograft tumor size in both the shRNA-control and trastuzumab-treated groups, the mean tumor size of the SLUG/SNAIL2 KD-JIMT1 cells was smaller (768 ± 145 mm3). Of note, when the SLUG/SNAIL2 KD-JIMT1 xenografts were treated with trastuzumab weekly, the mean tumor size was drastically reduced to 207 ± 76 mm3, thus showing that the trastuzumab-refractory JIMT1 xenografts gain sensitivity to trastuzumab when they are depleted of the EMT driver SLUG/SNAIL2. Consequently, approximately 25 d were required to observe a 10-fold increase in the tumor volume in the trastuzumab-refractory shRNA-control group, whereas more than 60 d were required when the EMT driver SLUG/SNAIL2 was depleted in the JIMT1 cells (, bottom).
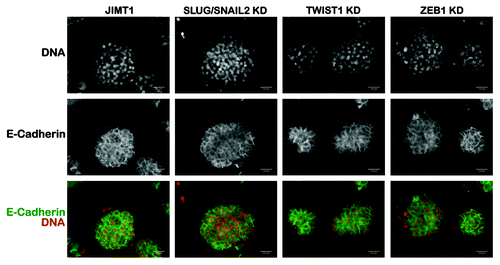
Loss of the SLUG/SNAIL2-driven CD44+CD24-/low mesenchymal subpopulation does not alter the mammosphere morphology of trastuzumab-refractory JIMT1 cells
The formation of primary mammospheres is a reliable in vitro assay to measure stem cell/early progenitor activity and, therefore, to demonstrate the presence of a stem cell population.Citation50-Citation53 Because CD44+CD24-/low cells have repeatedly been designated as highly tumorigenic breast CSCs,Citation3,Citation54-Citation56 we decided to establish primary mammospheres from CD44+CD24-/low-positive parental JIMT1 cells and their CD44+CD24-/low-negative derivatives, cells in which the expression of SLUG/SNAIL2 was stably knocked-down by a lentivirus-delivered shRNA. It is well-established that there are morphological differences in the mammospheres generated from different cell lines and different sources; e.g., mammospheres generated from CD44+C24-/low MDA-MB-231 mesenchymal cells form much looser structures of cell clumps compared with those derived from CD44-CD24+ MCF-7 luminal cells. We made the unexpected observation that, unlike the loose, grape-like structures generated by mesenchymal breast cancer cells,Citation57 the mammospheres generated from the CD44+C24-/low-enriched basal/HER2+ JIMT1 parental cells were solid structures with a rounded phenotype (). Therefore, these structures were essentially equivalent to the mammospheres generated from breast cancer cells bearing typical luminal epithelial features (e.g., T47D, MCF-7 or BT-474). Moreover, the mammospheres generated from the SLUG/SNAIL2 knockdown derivatives fully retained the compact and rounded morphology of the mammospheres generated by the parental JIMT1 cells and the expression of markers typical of the luminal/basal epithelial phenotype of breast cancer cells, such as E-cadherin (). The knockdowns of TWIST1 and ZEB1 also failed to alter mammosphere morphology in JIMT1 cells.
Figure 8. Mammosphere morphology of basal/HER2+ JIMT1 cells in response to the specific knockdown of SLUG/SNAIL2. The impact of shRNA-driven genetic ablation of SLUG/SNAIL2, TWIST1 and ZEB1 in mammosphere morphology was determined after seeding JIMT1 parental cells and SLUG/SNAIL2 KD-JIMT1, TWIST1 KD-JIMT1 and ZEB1 KD-JIMT1 cells in ultralow attachment plates with mammosphere medium for 5–7 d. Immunostaining of E-cadherin was performed on established spheres to visually evaluate whether the knockdown of EMT transcriptional drivers may result in disrupted mammosphere morphology. Genetic ablation of EMT drivers by lentivirus-delivered shRNAs did not affect mammosphere morphology of basal/HER2+ JIMT1 cells. KD, knockdown
Discussion
HER2 overexpression has been shown to represent a distinct tumor subtype in multiple independent cohorts of breast cancer patients; it is noteworthy, however, that most breast carcinoma-derived HER2+ cell lines cluster into the luminal subtype. Interestingly, although basal-like tumors most resemble basal A cell lines, and luminal-A and -B tumors most resemble luminal cell lines, the HER2+ tumor subgroup most resembles either the luminal or basal A cell lines. The findings presented here strongly suggest that the molecular basis for the discrepant HER2 grouping in cell lines and tumors may also explain the intrinsic resistance of HER2-positive breast carcinomas to trastuzumab. Thus, although the transcriptional and genomic profiles support the conclusion that the luminal and basal A cell lines are the most appropriate cell line models of luminal-B and basal-like tumors, respectively, we now provide experimental evidence to support a scenario in which the relative enrichment of EMT features that naturally occurs in basal-like breast cancer cells may indicate a new subgroup of HER2 gene-amplified breast carcinomas (basal/HER2+) with a primary resistance to HER2-targeted therapies, such as trastuzumab. In this scenario, the SLUG/SNAIL2-driven CD44+CD24-/low mesenchymal immunophenotype in the HER2 gene-amplified genomic background may induce an enhanced phenotypic plasticity that would allow the basal/HER2+ breast cancer cells to “enter” into and “exit” dynamically from HER2-driven stem cell-like states, which are primarily expected to be sensitive to trastuzumab.Citation1,Citation16
It is well-established that some members of the SNAIL family (i.e., SLUG/SNAIL2) and TWIST can confer an EMT phenotype to breast epithelial cells, a phenomenon that generally correlates with the cells changing their phenotype from CD44-CD24+ to CD44+CD24-/low.Citation58-Citation62 We now confirm the possibility that the EMT drivers SLUG/SNAIL2 and TWIST can also participate in the conversion of CD44+CD24+ basal epithelial cells into CD44+CD24-/low mesenchymal cells. More importantly, the fact that the HER2+ basal epithelial cells are susceptible to such EMT-related phenotypic changes can explain the intrinsic ability of a particular subgroup of HER2 overexpressors to escape inherently from the growth-inhibitory actions of HER2-targeting drugs, including the anti-HER2 monoclonal antibody trastuzumab. Because increasing evidence indicates a link among basal-like tumors, the stem cell phenotype, EMT and the acquisition of tumorigenic, invasive and metastatic potential,Citation63 and because breast stem cells have been shown to display a basal-like phenotype with an increased expression of EMT-related genes (e.g., SLUG/SNAIL2, VIM and N-cadherin),Citation64-Citation69 it might be tempting to suggest that the primary resistance to trastuzumab in the basal/HER2 subtype can be explained in terms of an intrinsic enrichment of the CSCs with EMT features. Although this suggestion might appear counterintuitive, because trastuzumab-resistant basal/HER2 cells are positive for both markers (i.e., CD44+CD24+) and because of the widely accepted CD44+CD24-/low mesenchymal immunophenotype of breast CSCs, it should be noted that CD24 is dynamically regulated, as recently demonstrated by Meyer et al.Citation46 When we previously explored how trastuzumab-refractory basal/HER2+ JIMT1 cells inherently regulated the CD44+CD24-/low surface markers,Citation12 we observed that the dynamic expression of the EMT-related markers was not limited to CD44/CD24, i.e., the number of cells bearing the CD44+CD24-/low mesenchymal immunophenotype switched over time from 10% in early passages to 80% in late passages. This was because the JIMT1 cell cultures enriched with CD44+CD24-/low mesenchymal cells also exhibited a reduced expression of the HER2 protein and an increased secretion of pro-invasive/metastatic chemokines and metalloproteases.
If the special proclivity of basal/HER2+ cancer cells to undergo EMT reflects the intrinsic phenotypic plasticity of HER2+ CSCs, it might be reasonable to suggest that EMT inducers, such as SLUG/SNAIL2, significantly impact the primary responses to trastuzumab by regulating the acquisition of invasive properties and/or the differentiation status of CS-like cells in basal/HER2+ breast carcinomas. Indeed, our current findings provide a wider therapeutic perspective to a landmark study by Moody et al.,Citation70 which used a conditional transgenic mouse model of the recurrence of HER2-induced mammary tumors to demonstrate that SLUG/SNAIL2 can be spontaneously upregulated in recurrent tumors in vivo, and that recurrence is accompanied by EMT and HER2 downregulation. The depletion of the SLUG/SNAIL2-driven CD44+CD24-/low mesenchymal subpopulation reduces the tumorigenic potential of JIMT1 cells and switches the trastuzumab-refractory phenotype of the basal/HER2+ JIMT1 cells to a sensitive phenotype; additionally, the cells bearing the CD44+CD24-/low surface phenotype have repeatedly been designated as the CSC subpopulation of breast cancer cells that is enriched in tumorigenic cells.Citation3,Citation54-Citation56 Accordingly, it might be reasonable to conclude that the aberrant expression of the EMT transcription factor SLUG/SNAIL2 biases HER2+ basal epithelial cells toward a highly tumorigenic CSC-like mesenchymal fate that is unresponsive to trastuzumab. Alternatively, the CSCs may not be considered distinct entities, but rather tumor cells that transiently acquire or lose stem cell-like properties as a consequence of EMT regulation. Although CD44+CD24-/low cells were originally described as CSCs, it is possible that the CSCs from the luminal/HER2+ cell types may be different from the basal/HER2+ cells types and may be characterized as CD44+CD24+, an intermediate phenotype expressing moderate levels of E-cadherin but expressing N-cadherin and VIM at similar levels as bona fide CD44+CD24-/low mesenchymal cells. Moreover, basal cell types but not luminal cell types may be susceptible to the SLUG/SNAIL2-driven acquisition of the CD44+CD24-/low mesenchymal phenotype, thus explaining both the intrinsic sensitivity to trastuzumab in luminal/HER2+ breast cancer cells and the intrinsic refractoriness to trastuzumab in basal/HER2+ breast cancer cells.
Although the results of this study highlight SLUG/SNAIL2 as an ideal candidate to induce the trastuzumab-refractory CD44+CD24-/low mesenchymal phenotype, it remains to be unambiguously defined whether the SLUG/SNAIL2-driven CD44+CD24-/low subpopulation reflects an enrichment of trastuzumab-refractory CSC mesenchymal cells or, alternatively, an intrinsic mechanism of escape from more epithelial, trastuzumab-responsive CS-like cellular states. Celià-Terrasa et al.Citation71 have recently proposed that highly metastatic tumor-initiating cells are enriched for a strong epithelial gene program, whereas tumor cell subpopulations with stable mesenchymal traits are notably depleted in CSCs. In their study, the constitutive overexpression of EMT transcription factors (i.e., SNAIL1) in epithelial CSC-enriched populations fully suppressed their self-renewal and metastatic phenotypes upon the activation of a mesenchymal gene program; conversely, the knockdown of EMT factors in the mesenchymal subpopulation caused an increase in the CSC-like properties, along with an enhancement of the epithelial features. It should be noted that the generation of induced pluripotent stem cells (iPSCs) from normal adult fibroblasts requires the proper activation and functioning of the mesenchymal-to-epithelial transition (MET) program, which is orchestrated by suppressing pro-EMT signals from the culture medium (e.g., the EMT activator TGFβ) and activating an epithelial gene program intracellularly.Citation72-Citation76 Accordingly, it has been shown that the expression of only E-cadherin can facilitate the reprogramming of adult fibroblasts and the acquisition of pluripotency.Citation75,Citation76 In fact, the four canonical Yamanaka pluripotency factors transcriptionally impede the EMT process: Sox2/Oct4 suppress the EMT mediator SNAIL; c-Myc downregulates TGF β1 and TGF β receptor 2 and Klf4 induces epithelial genes, including E-cadherin.Citation72-Citation77 Experiments are currently underway in our laboratory to determine whether the gain of epithelial features (e.g., E-cadherin expression) upon the silencing of EMT drivers in basal/HER2+ JIMT1 cells relates to changes in the endogenous expression of pluripotent transcription factors. Considering that bona fide mammospheres are non-adherent spherical cell clusters enriched in mammary stem/progenitor cells, it is intriguing that the steady depletion of CD44+CD24-/low mesenchymal cells in response to the specific knockdown of SLUG/SNAIL2 does not affect the epithelial nature of the mammospheres generated by the trastuzumab-sensitive SLUG/SNAIL2 KD-JIMT1 cells. Our findings favor a functional heterogeneity in the breast CS-like compartment and suggest that, in addition to the widely recognized CSCs with mesenchymal-like phenotypes, tumor-initiating cells with an epithelia-like morphology and functionality might be considered to understand better the breast cancer responses to molecularly targeted drugs. Preliminary observations in our laboratory strongly suggest that luminal epithelial breast cancer cells cannot be reprogrammed to pluripotent stem cell states upon the silencing of E-cadherin, whereas HER2 expression appears to be upregulated when fibroblasts, a product of EMT, are pushed to an E-cadherin-positive stem cell-like state via exogenous transcription factors (data not shown). Unpublished observations by Wichas’s group indicate that clinically HER2-negative primary luminal breast carcinomas contain a CS-like subpopulation of cells that express the HER2 protein on the invasive edge of the tumor.Citation78 Although this pattern can explain why HER2-normal patients benefit from trastuzumab, it is noteworthy that basal-type tumors fail to induce trastuzumab-sensitive HER2+ CS-like cells, once again linking trastuzumab efficacy with the epithelial behavior of breast CSCs.
Using trastuzumab monotherapy as an example, Neve et al.Citation39 concluded that the gene expression data sets from the 51 luminal, basal A and basal B (mesenchymal) breast cancer cell lines in their system can be used to identify accurately the molecular features that predict a response to targeted therapies. By following a similar approach, we now confirm that, when HER2 gene amplification, which generally occurs in differentiated luminal phenotypes, occurs in a basal-like molecular background, it results in a basal/HER2+ breast cancer subtype that is inherently resistant to trastuzumab. By molecularly tracking the effectors of the de novo unresponsiveness of the basal/HER2+ cells to trastuzumab, we concluded that the expression of EMT-driving transcriptional factors was causally related to the trastuzumab resistance phenotype. The EMT-driving transcriptional repressor SLUG/SNAIL2 appears to be a pivotal gene that induces an enhanced phenotypic plasticity in basal/HER2+ cells, thus allowing them to “enter” into and “exit” from trastuzumab-responsive CS-like states. In this previously unrecognized scenario, our results encourage the systematic determination of SLUG/SNAIL2 as a stem/CD44+CD24-/low cell-associated protein to provide better therapeutic management of HER2 gene-amplified breast carcinomas. The targeting of the EMT phenomenon is, therefore, a promising therapeutic strategy to prevent and circumvent the de novo resistance to trastuzumab-based therapeutic regimens.Citation36,Citation79-Citation87
Materials and Methods
Culture conditions
Parental JIMT1 breast carcinoma cells were obtained from the German Collection of Microorganisms. The cells were routinely grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco® Cell Culture Systems, Invitrogen S.A.) containing 10% heat-inactivated fetal bovine serum (FBS, Bio-Whittaker, Inc.), 1% L-glutamine, 1% sodium pyruvate, 50 U/mL penicillin and 50 μ/mL streptomycin. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. The cells were also periodically screened for Mycoplasma contamination. The JIMT1 cells stably expressing the small hairpin (sh) RNAs to knockdown (KD) the expression of SLUG/SNAIL2 (SLUG/SNAIL2 KD-JIMT1), TWIST1 (TWIST KD-JIMT1) or ZEB1 (ZEB KD-JIMT1) were maintained in puromycin-containing media.
Drugs
Trastuzumab (Herceptin®) was kindly provided by the Hospital Universitari de Girona Dr. Josep Trueta Pharmacy. Trastuzumab was solubilized in bacteriostatic water containing 1.1% benzyl alcohol (stock solution at 21 mg/mL for injection, stored at 4 þC and used within 1 mo). For experimental use, trastuzumab was prepared fresh from stock solutions and diluted with the cell growth medium immediately before use.
Stable silencing of EMT transcription factors
Pre-packaged lentiviral particles were purchased from a commercial provider (Santa Cruz Biotechnology) and encoded either a non-targeting shRNA (negative shRNA, sc-108080) or sequences specifically targeting the human SLUG/SNAIL2, TWIST1 or ZEB1 genes. For the viral infection of the JIMT1 cells, the regular medium was replaced with a culture medium containing 5 mg/mL polybrene (Santa Cruz Biotechnology, sc-124220). The JIMT1 cells were then exposed to lentiviruses for 48 h. Because the lentiviral shRNA particles also encode a puromycin resistance gene for the transduction selection, the cells were washed and grown in a culture medium containing 10 μg/mL puromycin dihydrochloride (Sigma, Cat. No. P9620) for an additional 72 h. The JIMT1 cells were allowed to recover and proliferate for at least 1 wk prior to any experimental procedure and were then processed for the analyses. To monitor the lentiviral transduction efficiency and transgene expression for the duration of the experiments, we incubated additional subsets of JIMT1 cells with lentiviral particles encoding a green fluorescence protein (GFP) reporter (Santa Cruz Biotechnology, sc-108084). The transduction efficiency (> 90%) was determined as the ratio of the number of GFP-positive cells to the total number of cells in five random visual fields obtained from three independent culture experiments.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The total RNA was extracted from cell cultures using a Qiagen RNeasy kit and QIAshredder columns according to the manufacturer’s instructions. One microgram of total RNA was reverse-transcribed to cDNA using the Reaction Ready™ First Strand cDNA Synthesis Kit (SABiosciences) and applied to a customized PCR array (96-well format) containing the following panel of genes: GSC, KRT14, KRT19, NUMB, TCF3, TCF4, SDC1, ZO-1, CD44, TWIST, SNAI1, VIM, SLUG, CDH1, ZEB1, CDH2, ZEB2, FN1 and CD24. The arrays were processed according to the SABiosciences RT-PCR manual and analyzed using an Applied Biosystems 7500 Fast Real-Time PCR System with an automated baseline and threshold cycle detection. The data were interpreted using SABiosciences’s web-based PCR array analysis tool.
Flow cytometry
The cells were washed once with phosphate-buffered saline (PBS) and then harvested with 0.05% trypsin/0.025% EDTA into single-cell suspensions. The detached cells were washed with PBS containing 1% FBS and 1% penicillin/streptomycin (wash buffer), counted and resuspended in the wash buffer (106 cells/100 μl). Combinations of fluorochrome-conjugated monoclonal antibodies (obtained from BD Biosciences) against CD44 (FITC conjugated, Cat. No. 555478) and CD24 (PE conjugated, Cat. No. 555428) or their respective isotype controls were added to the cell suspension at the concentrations recommended by the manufacturer and incubated at 4°C in the dark for 30–40 min. The labeled cells were washed in the wash buffer to eliminate unbound antibody, fixed in PBS containing 1% paraformaldehyde and analyzed no longer than 1 h post-staining using a BD FACSCalibur (BD Biosciences).
Cell proliferation assays
The cells were trypsinized and re-plated in 24-well plates at a density of 10,000 cells/well. The cells were incubated for 18 h to allow for attachment, after which a 0-time point measurement was determined. The cells were then cultured in the routine medium containing 5% FBS in the absence or presence of 100 μg/mL trastuzumab and counted at days 0, 3, 6 and 8 using a Coulter Counter (Coulter Electronics, Inc.). All of the assays were performed at least three times in duplicate.
Cell viability assays
The effect of trastuzumab on the cell viability was determined using a standard colorimetric MTT (3–4, 5-dimethylthiazol-2-yl-2, 5-diphenyl-tetrazolium bromide) reduction assay. For each treatment, the cell viability was evaluated as a percentage using the following equation: (OD570 of the treated sample/OD570 of the untreated sample) × 100.
Isolation of the CD44+CD24-/low mesenchymal immunophenotype using MACS technology
The JIMT1 cells were sorted by sequential sorting (depletion, followed by positive selection) using MACS MicroBeads (MACS® Technology). The enrichment of the target cells with the magnetic MicroBeads was performed according to the manufacturer’s protocol (Milteny Biotec). The CD44+CD24-/low cells were isolated from the parental JIMT-1 cell line by first depleting the CD24+ cells using the CD24 MicroBead Kit and then positively selecting for CD44 using CD44 MicroBeads. The cells were fluorescently stained with combinations of fluorochrome-conjugated monoclonal antibodies obtained from BD Biosciences against human CD44 (FITC; cat.#555478) and CD24 (PE; cat.#555428) or their respective isotype controls. The figures show the representative expression (n = 5) in the pre-sorted and post-sorted JIMT1 cells.
Immunofluorescence staining and high-content confocal imaging
Cells were seeded at approximately 5,000 cells/well in 96-well clear-bottom imaging tissue culture plates (Becton Dickinson Biosciences) optimized for automated imaging applications. Triton® X-100 permeabilization and blocking, primary antibody staining, secondary antibody staining using Alexa Fluor® 488/594 goat anti-rabbit/mouse IgGs (Invitrogen, Probes) and counterstaining (using Hoechst 33258; Invitrogen) were performed following the BD Biosciences protocols. The images were captured in different channels for Alexa Fluor® 488 (pseudocolored green), Alexa Fluor® 594 (pseudocolored red) and Hoechst 33258 (pseudocolored blue) using a BD PathwayTM 855 Bioimager System (Becton Dickinson Biosciences) with 20x or 40x objectives (NA 075 Olympus). Merged images were obtained according to the Recommended Assay Procedure using the BD AttovisionTM software.
Xenograft studies
The Institutional Animal Care and Use Committee approved all animal studies. To produce xenografts, approximately 5 × 106 JIMT1 or SLUG/SNAIL2KD-JIMT1 cells were injected subcutaneously into the dorsal flank of female athymic nude mice (4–5 wk old, 23–25 g; Harlan Laboratories). The animals were randomized into two groups, with five animals in each group: control (vehicle) and trastuzumab treated. Trastuzumab (5 mg/kg) was administered intraperitoneally (i.p.) once per week. The mice were weighed once per week after dosing; the tumor sizes were measured daily using electronic calipers; and the tumor volumes were calculated using the following formula: volume (mm3) = length × width2 × 0.5. The figures show the mean tumor volumes (± SD) of the JIMT1 and SLUG/SNAIL2 KD-JIMT1 xenograft-bearing nude mice following the injections with trastuzumab for 9 wk.
Generation of mammospheres
For mammosphere formation, single-cell suspensions were plated in 6-well tissue culture plates coated with poly-2-hydroxyethyl-methacrylate (Sigma) to prevent cell attachment at a density of 1,000 cells/mL in serum-free DMEM supplemented with 1% L-glutamine, 1% penicillin/streptomycin, 30% F12 (Sigma), 2% B27 (Invitrogen), 20 ng/mL EGF (Sigma) and 20 ng/mL FGFb (Invitrogen). The medium was made semi-solid through the addition of 0.5% methylcellulose (R&D Systems) to prevent cell aggregation. After 7 d in culture, mammospheres were collected by gentle centrifugation (150 × g) and dissociated enzymatically (5 min in 1:1 trypsin/DMEM solution at 37°C) and mechanically by passaging through a 25 G needle (6 strokes). The single cells were re-plated at a density of 1,000 cells/mL for subsequent passages.
Acknowledgments
This work was financially supported by the Instituto de Salud Carlos III [Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain, grants CP05–00090, PI06–0778 and RD06–0020–0028], the Fundación Científica de la Asociación Española Contra el Cáncer (AECC, Spain) and the Ministerio de Ciencia e Innovación (SAF2009–11579, Plan Nacional de I+D+ I, MICINN, Spain). A.V.-M. received the Sara Borrell post-doctoral contract (CD08/00283, Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria -FIS-, Spain). S.C. received a research fellowship (Formación de Personal Investigador, FPI) from the Ministerio de Ciencia e Innovación (MICINN, Spain).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008; 27:6120 - 30; http://dx.doi.org/10.1038/onc.2008.207; PMID: 18591932
- Kakarala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol 2008; 26:2813 - 20; http://dx.doi.org/10.1200/JCO.2008.16.3931; PMID: 18539959
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983 - 8; http://dx.doi.org/10.1073/pnas.0530291100; PMID: 12629218
- Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target?. J Natl Cancer Inst 2004; 96:583 - 5; http://dx.doi.org/10.1093/jnci/djh095; PMID: 15100335
- Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res 2008; 10:R25; http://dx.doi.org/10.1186/bcr1982; PMID: 18366788
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100:672 - 9; http://dx.doi.org/10.1093/jnci/djn123; PMID: 18445819
- Debeb BG, Xu W, Woodward WA. Radiation resistance of breast cancer stem cells: understanding the clinical framework. J Mammary Gland Biol Neoplasia 2009; 14:11 - 7; http://dx.doi.org/10.1007/s10911-009-9114-z; PMID: 19252973
- Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008; 26:2839 - 45; http://dx.doi.org/10.1200/JCO.2007.15.1829; PMID: 18539962
- Nguyen NP, Almeida FS, Chi A, Nguyen LM, Cohen D, Karlsson U, et al. Molecular biology of breast cancer stem cells: potential clinical applications. Cancer Treat Rev 2010; 36:485 - 91; http://dx.doi.org/10.1016/j.ctrv.2010.02.016; PMID: 20231058
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med 2007; 356:217 - 26; http://dx.doi.org/10.1056/NEJMoa063994; PMID: 17229949
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA 2009; 106:13820 - 5; http://dx.doi.org/10.1073/pnas.0905718106; PMID: 19666588
- Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castillo B, Cufí S, Del Barco S, Lopez-Bonet E, et al. Dynamic emergence of the mesenchymal CD44(pos)CD24(neg/low) phenotype in HER2-gene amplified breast cancer cells with de novo resistance to trastuzumab (Herceptin). Biochem Biophys Res Commun 2010; 397:27 - 33; http://dx.doi.org/10.1016/j.bbrc.2010.05.041; PMID: 20470755
- Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat 2011; 126:355 - 64; http://dx.doi.org/10.1007/s10549-010-0924-x; PMID: 20458531
- Jiang J, Zhang Y, Chuai S, Wang Z, Zheng D, Xu F, et al. Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene 2012; 31:671 - 82; http://dx.doi.org/10.1038/onc.2011.282; PMID: 21743497
- Liu JC, Voisin V, Bader GD, Deng T, Pusztai L, Symmans WF, et al. Seventeen-gene signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2+:ERα- breast cancer. Proc Natl Acad Sci USA 2012; 109:5832 - 7; http://dx.doi.org/10.1073/pnas.1201105109; PMID: 22460789
- Magnifico A, Albano L, Campaner S, Delia D, Castiglioni F, Gasparini P, et al. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin Cancer Res 2009; 15:2010 - 21; http://dx.doi.org/10.1158/1078-0432.CCR-08-1327; PMID: 19276287
- Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med 2008; 358:1409 - 11; http://dx.doi.org/10.1056/NEJMc0801440; PMID: 18367751
- Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol 2010; 28:4006 - 12; http://dx.doi.org/10.1200/JCO.2009.27.5388; PMID: 20498387
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 2002; 20:719 - 26; http://dx.doi.org/10.1200/JCO.20.3.719; PMID: 11821453
- Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol 2006; 3:269 - 80; http://dx.doi.org/10.1038/ncponc0509; PMID: 16683005
- Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res 2006; 8:215; http://dx.doi.org/10.1186/bcr1612; PMID: 17096862
- Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol 2007; 18:977 - 84; http://dx.doi.org/10.1093/annonc/mdl475; PMID: 17229773
- Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene 2007; 26:3637 - 43; http://dx.doi.org/10.1038/sj.onc.1210379; PMID: 17530017
- Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol 2009; 27:5838 - 47; http://dx.doi.org/10.1200/JCO.2009.22.1507; PMID: 19884552
- Esteva FJ, Yu D, Hung MC, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol 2010; 7:98 - 107; http://dx.doi.org/10.1038/nrclinonc.2009.216; PMID: 20027191
- Pályi-Krekk Z, Barok M, Isola J, Tammi M, Szöllosi J, Nagy P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur J Cancer 2007; 43:2423 - 33; http://dx.doi.org/10.1016/j.ejca.2007.08.018; PMID: 17911008
- Pályi-Krekk Z, Barok M, Kovács T, Saya H, Nagano O, Szöllosi J, et al. EGFR and ErbB2 are functionally coupled to CD44 and regulate shedding, internalization and motogenic effect of CD44. Cancer Lett 2008; 263:231 - 42; http://dx.doi.org/10.1016/j.canlet.2008.01.014; PMID: 18276068
- Dawood S, Gong Y, Broglio K, Buchholz TA, Woodward W, Lucci A, et al. Trastuzumab in Primary Inflammatory Breast Cancer (IBC): High Pathological Response Rates and Improved Outcome. Breast J 2010; PMID: 20626396
- Bao W, Fu HJ, Xie QS, Wang L, Zhang R, Guo ZY, et al. HER2 interacts with CD44 to up-regulate CXCR4 via epigenetic silencing of microRNA-139 in gastric cancer cells. Gastroenterology 2011; 141:2076 - 87, e6; PMID: 21925125
- Lesniak D, Xu Y, Deschenes J, Lai R, Thoms J, Murray D, et al. Beta1-integrin circumvents the antiproliferative effects of trastuzumab in human epidermal growth factor receptor-2-positive breast cancer. Cancer Res 2009; 69:8620 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-09-1591; PMID: 19887601
- Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene 2008; 27:5019 - 32; http://dx.doi.org/10.1038/onc.2008.149; PMID: 18469855
- Pandya K, Meeke K, Clementz AG, Rogowski A, Roberts J, Miele L, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer 2011; 105:796 - 806; http://dx.doi.org/10.1038/bjc.2011.321; PMID: 21847123
- Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Torres-Garcia VZ, Sauri-Nadal T, Barco SD, et al. Inhibitor of Apoptosis (IAP) survivin is indispensable for survival of HER2 gene-amplified breast cancer cells with primary resistance to HER1/2-targeted therapies. Biochem Biophys Res Commun 2011; 407:412 - 9; http://dx.doi.org/10.1016/j.bbrc.2011.03.039; PMID: 21402055
- Bedard PL, Cardoso F, Piccart-Gebhart MJ. Stemming resistance to HER-2 targeted therapy. J Mammary Gland Biol Neoplasia 2009; 14:55 - 66; http://dx.doi.org/10.1007/s10911-009-9116-x; PMID: 19259796
- Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castilló B, Pérez-Martínez MC, Cufí S, Del Barco S, et al. Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotype: new insights into de novo resistance to trastuzumab (Herceptin). Int J Oncol 2010; 37:669 - 78; PMID: 20664936
- Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, et al. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012; 3:395 - 8; PMID: 22565037
- Blick T, Widodo E, Hugo H, Waltham M, Lenburg ME, Neve RM, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis 2008; 25:629 - 42; http://dx.doi.org/10.1007/s10585-008-9170-6; PMID: 18461285
- Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia 2010; 15:235 - 52; http://dx.doi.org/10.1007/s10911-010-9175-z; PMID: 20521089
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10:515 - 27; http://dx.doi.org/10.1016/j.ccr.2006.10.008; PMID: 17157791
- Tanner M, Kapanen AI, Junttila T, Raheem O, Grenman S, Elo J, et al. Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol Cancer Ther 2004; 3:1585 - 92; PMID: 15634652
- Nagy P, Friedländer E, Tanner M, Kapanen AI, Carraway KL, Isola J, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res 2005; 65:473 - 82; PMID: 15695389
- Jönsson G, Staaf J, Olsson E, Heidenblad M, Vallon-Christersson J, Osoegawa K, et al. High-resolution genomic profiles of breast cancer cell lines assessed by tiling BAC array comparative genomic hybridization. Genes Chromosomes Cancer 2007; 46:543 - 58; http://dx.doi.org/10.1002/gcc.20438; PMID: 17334996
- Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res 2011; 71:245 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-10-2330; PMID: 21199805
- Vesuna F, Lisok A, Kimble B, Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009; 11:1318 - 28; PMID: 20019840
- Bhat-Nakshatri P, Appaiah H, Ballas C, Pick-Franke P, Goulet R Jr., Badve S, et al. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24- phenotype. BMC Cancer 2010; 10:411; http://dx.doi.org/10.1186/1471-2407-10-411; PMID: 20691079
- Meyer MJ, Fleming JM, Ali MA, Pesesky MW, Ginsburg E, Vonderhaar BK. Dynamic regulation of CD24 and the invasive, CD44posCD24neg phenotype in breast cancer cell lines. Breast Cancer Res 2009; 11:R82; http://dx.doi.org/10.1186/bcr2449; PMID: 19906290
- Kaipparettu BA, Malik S, Konduri SD, Liu W, Rokavec M, van der Kuip H, et al. Estrogen-mediated downregulation of CD24 in breast cancer cells. Int J Cancer 2008; 123:66 - 72; http://dx.doi.org/10.1002/ijc.23480; PMID: 18404683
- Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 2002; 62:1613 - 8; PMID: 11912130
- Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 2003; 116:499 - 511; http://dx.doi.org/10.1242/jcs.00224; PMID: 12508111
- Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res 2005; 65:5506 - 11; http://dx.doi.org/10.1158/0008-5472.CAN-05-0626; PMID: 15994920
- Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif 2003; 36:Suppl 1 59 - 72; http://dx.doi.org/10.1046/j.1365-2184.36.s.1.6.x; PMID: 14521516
- Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res 2010; 70:709 - 18; http://dx.doi.org/10.1158/0008-5472.CAN-09-1681; PMID: 20068161
- Shaw FL, Harrison H, Spence K, Ablett MP, Simões BM, Farnie G, et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia 2012; 17:111 - 7; http://dx.doi.org/10.1007/s10911-012-9255-3; PMID: 22665270
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704 - 15; http://dx.doi.org/10.1016/j.cell.2008.03.027; PMID: 18485877
- Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008; 3:e2888; http://dx.doi.org/10.1371/journal.pone.0002888; PMID: 18682804
- Floor S, van Staveren WC, Larsimont D, Dumont JE, Maenhaut C. Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: distinct, overlapping or same populations. Oncogene 2011; 30:4609 - 21; http://dx.doi.org/10.1038/onc.2011.184; PMID: 21643013
- Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010; 9:3807 - 14; http://dx.doi.org/10.4161/cc.9.18.13131; PMID: 20890129
- Alves CC, Carneiro F, Hoefler H, Becker KF. Role of the epithelial-mesenchymal transition regulator Slug in primary human cancers. Front Biosci 2009; 14:3035 - 50; http://dx.doi.org/10.2741/3433; PMID: 19273255
- Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 2010; 15:117 - 34; http://dx.doi.org/10.1007/s10911-010-9178-9; PMID: 20490631
- Hugo HJ, Kokkinos MI, Blick T, Ackland ML, Thompson EW, Newgreen DF. Defining the E-cadherin repressor interactome in epithelial-mesenchymal transition: the PMC42 model as a case study. Cells Tissues Organs 2011; 193:23 - 40; http://dx.doi.org/10.1159/000320174; PMID: 21051859
- Foubert E, De Craene B, Berx G. Key signalling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res 2010; 12:206; http://dx.doi.org/10.1186/bcr2585; PMID: 20594364
- Kang Y, Massagué J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 2004; 118:277 - 9; http://dx.doi.org/10.1016/j.cell.2004.07.011; PMID: 15294153
- Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, et al. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol 2007; 213:374 - 83; http://dx.doi.org/10.1002/jcp.21223; PMID: 17680632
- Storci G, Sansone P, Trere D, Tavolari S, Taffurelli M, Ceccarelli C, et al. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J Pathol 2008; 214:25 - 37; http://dx.doi.org/10.1002/path.2254; PMID: 17973239
- Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer 2007; 7:791 - 9; http://dx.doi.org/10.1038/nrc2212; PMID: 17851544
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007; 11:259 - 73; http://dx.doi.org/10.1016/j.ccr.2007.01.013; PMID: 17349583
- Liao MJ, Zhang CC, Zhou B, Zimonjic DB, Mani SA, Kaba M, et al. Enrichment of a population of mammary gland cells that form mammospheres and have in vivo repopulating activity. Cancer Res 2007; 67:8131 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-4493; PMID: 17804725
- Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res 2008; 68:989 - 97; http://dx.doi.org/10.1158/0008-5472.CAN-07-2017; PMID: 18281472
- Gasparotto D, Polesel J, Marzotto A, Colladel R, Piccinin S, Modena P, et al. Overexpression of TWIST2 correlates with poor prognosis in head and neck squamous cell carcinomas. Oncotarget 2011; 2:1165 - 75; PMID: 22201613
- Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005; 8:197 - 209; http://dx.doi.org/10.1016/j.ccr.2005.07.009; PMID: 16169465
- Celià-Terrassa T, Meca-Cortés O, Mateo F, de Paz AM, Rubio N, Arnal-Estapé A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest 2012; 122:1849 - 68; http://dx.doi.org/10.1172/JCI59218; PMID: 22505459
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010; 7:51 - 63; http://dx.doi.org/10.1016/j.stem.2010.04.014; PMID: 20621050
- Polo JM, Hochedlinger K. When fibroblasts MET iPSCs. Cell Stem Cell 2010; 7:5 - 6; http://dx.doi.org/10.1016/j.stem.2010.05.018; PMID: 20621040
- Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010; 7:64 - 77; http://dx.doi.org/10.1016/j.stem.2010.04.015; PMID: 20621051
- Lowry WE. E-cadherin, a new mixer in the Yamanaka cocktail. EMBO Rep 2011; 12:613 - 4; http://dx.doi.org/10.1038/embor.2011.117; PMID: 21701504
- Redmer T, Diecke S, Grigoryan T, Quiroga-Negreira A, Birchmeier W, Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep 2011; 12:720 - 6; http://dx.doi.org/10.1038/embor.2011.88; PMID: 21617704
- Salem AF, Bonuccelli G, Bevilacqua G, Arafat H, Pestell RG, Sotgia F, et al. Caveolin-1 promotes pancreatic cancer cell differentiation and restores membranous E-cadherin via suppression of the epithelial-mesenchymal transition. Cell Cycle 2011; 10:3692 - 700; http://dx.doi.org/10.4161/cc.10.21.17895; PMID: 22041584
- Tuma RS. Cancer stem cell hypothesis and trastuzumab in HER2-negative tumors. J Natl Cancer Inst 2012; 104:968 - 9; http://dx.doi.org/10.1093/jnci/djs307; PMID: 22745473
- Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle 2011; 10:2959 - 66; http://dx.doi.org/10.4161/cc.10.17.16359; PMID: 21862872
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29:4741 - 51; http://dx.doi.org/10.1038/onc.2010.215; PMID: 20531305
- Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010; 9:3807 - 14; http://dx.doi.org/10.4161/cc.9.18.13131; PMID: 20890129
- Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S, et al. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist Updat 2010; 13:109 - 18; http://dx.doi.org/10.1016/j.drup.2010.07.001; PMID: 20692200
- Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle 2010; 9:4461 - 8; http://dx.doi.org/10.4161/cc.9.22.14048; PMID: 21088486
- Takebe N, Warren RQ, Ivy SP. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res 2011; 13:211; http://dx.doi.org/10.1186/bcr2876; PMID: 21672282
- Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin: multi-faceted protection against cancer. Oncotarget 2011; 2:896 - 917; PMID: 22203527
- Rattan R, Ali Fehmi R, Munkarah A. Metformin: an emerging new therapeutic option for targeting cancer stem cells and metastasis. J Oncol 2012; 2012:928127; PMID: 22701483
- Barrière G, Tartary M, Rigaud M. Metformin: A Rising Star to Fight The Epithelial Mesenchymal Transition in Oncology. Anticancer Agents Med Chem 2012; In press