Abstract
Krüppel-like factor 5 (KLF5) is a key transcriptional regulator that is typically pro-proliferative in non-transformed epithelial cells but inhibits proliferation in transformed epithelial cells. However, the underlying mechanisms for this context-dependent function are not known. KLF5 is epigenetically silenced and exhibits a tumor suppressive function in esophageal squamous cell cancer (ESCC). Since p53 mutation is the most common genetic alteration in ESCC, as in other human epithelial cancers, we hypothesized that the context-dependent functions of KLF5 in cell proliferation were dependent on p53 status. In fact, in non-transformed human primary esophageal keratinocytes, when p53 was wild-type, KLF5 was pro-proliferative; however, KLF5 became anti-proliferative when p53 was mutated. KLF5 loss in human primary keratinocytes harboring p53 mutation accelerated the cell cycle and decreased expression of p21Waf1/Cip1; similar effects were also seen in ESCC cells with established p53 mutations. Further, p21Waf1/Cip1 was directly and differentially bound and regulated by KLF5 in the presence or absence of mutant p53, and suppression of p21Waf1/Cip1 reversed the antiproliferative effects of KLF5 in the presence of p53 mutation. Thus, KLF5 is a critical brake on an aberrant cell cycle, with important tumor suppressive functions in esophageal squamous cell and potentially other epithelial cancers.
Keywords: :
Introduction
Coordination of gene transcription within epithelial cells is crucial for development, differentiation and tumorigenesis in the gastrointestinal tract and other tissues.Citation1,Citation2 Careful modulation of gene expression within these epithelia is required for normal growth control, and a number of key transcriptional regulators can play differing roles in these processes, depending on context and/or tissue type.Citation3,Citation4 For example, both loss and increased expression of the zinc-finger transcription factor Krüppel-like factor 4 (KLF4) alter normal esophageal and epidermal homeostasis.Citation5-Citation8 In breast cancer, this context-dependent role of KLF4 is linked to p21Waf1/Cip1, although the mediators of these effects in other epithelia are more complex.Citation5,Citation9 Notch1 is required for normal epithelial homeostasis and can function during epithelial carcinogenesis as either an oncogene or a tumor suppressor, the latter in the squamous epithelia of both skin and esophagus.Citation10-Citation12 Identifying the mediators of the context-dependent functions of these and other transcriptional regulators provides insight into the mechanisms of normal development, homeostasis, injury repair, regeneration and carcinogenesis.
Many epithelial transcriptional regulators have specific patterns of expression which are consistent with their typical functions.Citation2 Illustrative of this, the transcription factor Krüppel-like factor 5 (KLF5), which is frequently pro-proliferative, including in murine esophageal epithelia, is normally restricted to regions of active cell proliferation, such as the basal layer of the esophagus.Citation13-Citation15 In some contexts, KLF5 can upregulate cyclin B1 and cyclin D1,Citation16,Citation17 enhancing proliferation. Yet, KLF5 is growth inhibitory in other contexts, including in transformed intestinal cells and esophageal cancer cells.Citation18,Citation19 A clue to the mechanisms of this context-dependent function for KLF5 in epithelial cell proliferation may come from studies of the coordinate regulation of the Notch1 gene by KLF5 and p53 during esophageal squamous cell transformation.Citation20 In this instance, KLF5 and p53 bind within the same region on the Notch1 promoter in human primary esophageal keratinocytes; binding of either p53 or KLF5 results in transcriptional activation of Notch1 to inhibit cellular transformation, defining KLF5 as an esophageal tumor suppressor. Importantly, p53 mutation is a signature early event in squamous esophageal cancer,Citation21,Citation22 meaning that p53 status typically varies between normal esophageal epithelia cells and esophageal squamous cell cancer. Thus, changes in p53 status could be key for the context-dependent effects of KLF5. Still, the exact mechanisms for the differential effects of KLF5 on proliferation in normal homeostasis and cancer are not clear.
The human primary esophageal keratinocyte cell line EPC2-hTERT (EPC2-T) provides an important and relevant model for studies of the transcriptional regulation of proliferation, since these non-transformed epithelial cells lack the genetic modifications of both cancer cells and most spontaneously immortalized epithelial cell lines.Citation23,Citation24 As such, EPC2-T cells provide an ideal system for studies of the mechanisms of the context-dependent effects of KLF5 in normal epithelial homeostasis. Notably, EPC2-T cells express wild-type p53, in contrast to most human esophageal squamous cell cancer lines where p53 is either mutated or epigenetically silenced.Citation25 Here, we have utilized EPC2-T cells with or without the p53 hotspot mutation p53R175H, which is the most common mutation in esophageal squamous cell cancer and numerous other cancers,Citation26,Citation27 to demonstrate that KLF5 effects on proliferation are dependent on p53 status and are mediated specifically by p21Waf1/Cip1. In addition, we show that the cyclin-dependent kinase inhibitor p21Waf1/Cip1, like Notch1,Citation20 is coordinately regulated by KLF5 and p53. These results help to define the mechanisms of context-dependent, cell autonomous regulation of cellular proliferation during epithelial homeostasis and carcinogenesis.
Results
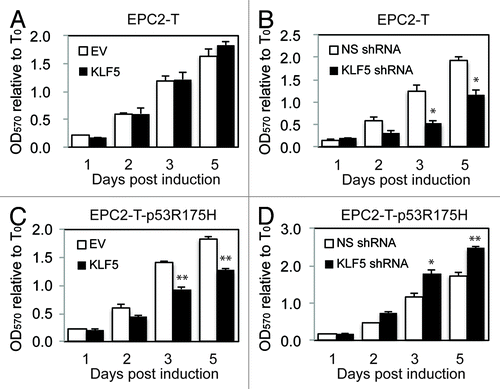
To investigate whether the function of KLF5 in cell proliferation is dependent on p53 status, we overexpressed, or knocked-down, KLF5 using an inducible system in human primary esophageal epithelial cells (EPC2-T) containing wild-type or mutant p53 (p53R175H).Citation20 Of note, p53R175H is one of the most common mutations in numerous epithelial cancers, including ESCC.Citation27 In EPC2-T cells, proliferation was not altered by KLF5 induction (), likely due to the presence of endogenous KLF5 in EPC2-T cells,Citation20 but proliferation was significantly decreased by KLF5 suppression (), confirming a growth-promoting role for KLF5 in the presence of wild-type p53. In contrast, KLF5 induction in EPC2-T-p53R175H cells resulted in a significant reduction in cell proliferation (), and proliferation was increased by KLF5-knockdown in EPC2-T-p53R175H cells (), indicating that KLF5 in this context had a growth-inhibitory effect. By flow cytometry, KLF5 suppression in EPC2-T-p53R175H cells decreased the number of cells in G2/M, with a corresponding increase in the number of cells in G1/S (Fig. S1A). No changes in apoptosis were seen with KLF5 suppression in EPC2-T cells or KLF5 induction in EPC2-T-p53R175H cells (Fig. S1B).
Figure 1. KLF5 promotes proliferation in cells with wild-type p53 but inhibits cell proliferation with mutant p53. (A and B) In EPC2-T cells, by MTT assay, KLF5 overexpression had little effect on cell proliferation (A), likely due to the effects of endogenous KLF5, but proliferation was significantly reduced by KLF5 suppression with shRNA (B). (C and D) In contrast, in EPC2-T-p53R175H cells, KLF5 induction inhibited cell proliferation (C), while KLF5 knockdown increased proliferation (D). EV, empty vector control; * p < 0.05, ** p < 0.005
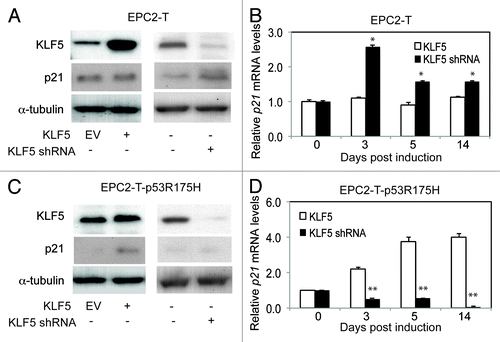
The cyclin-dependent kinase inhibitor p21Waf1/Cip1 is critical for progression through numerous cell cycle checkpoints, including the G1/S and G2/M transitions,Citation28-Citation30 and is a well-defined p53 target.Citation30,Citation31 In EPC2-T cells, KLF5 suppression resulted in increased p21Waf1/Cip1 expression (). KLF5 induction in EPC2-T-p53R175H cells, which similarly inhibits cell proliferation, also upregulated p21Waf1/Cip1 (), and knockdown of p21Waf1/Cip1 in EPC2-T-p53R175H cells produced a similar effect on the cell cycle as KLF5 suppression (Fig. S1C). To confirm the specificity for p21Waf1/Cip1, we examined the effects of KLF5 on the expression of the cyclin-dependent kinase inhibitors p15, p16, p18 and p27 (Fig. S2). While KLF5 suppression appeared to initially induce p15 and p27 differentially in EPC2-T cells and EPC2-T-p53R175H cells, this effect was not maintained over time, in contrast to the effect on p21Waf1/Cip1, and no changes were seen for p16 or p18. In the ESCC cell line TE7, both KLF5 and p53 are normally undetectable,Citation20,Citation25 and KLF5 expression also upregulated p21Waf1/Cip1 (Fig. S3A). These findings are consistent with a role for p21Waf1/Cip1 in mediating the effects of KLF5 on cell proliferation. Yet the mechanisms by which changes in p53 status might lead to differential effects of KLF5 on p21Waf1/Cip1 expression are not clear.
Figure 2. KLF5 upregulates the cyclin-dependent kinase inhibitor p21Waf1/Cip1 in cells with mutant p53. (A and B) When KLF5 was induced in EPC2-T cells, which contain wild-type p53, p21Waf1/Cip1 protein (A), by western blot, and mRNA (B), by quantitative real-time PCR, were unchanged, similar to the effects on proliferation seen with KLF5 induction. In contrast, KLF5 knockdown by shRNA, increased p21Waf1/Cip1 in EPC2-T cells. (C and D) In EPC2-T-p53R175H cells, p21Waf1/Cip1 was normally expressed at low levels, and KLF5 overexpression increased p21Waf1/Cip1 protein (C) and mRNA (D), while KLF5 suppression reduced p21Waf1/Cip1. EV, empty vector control; *p < 0.05, ** p < 0.01
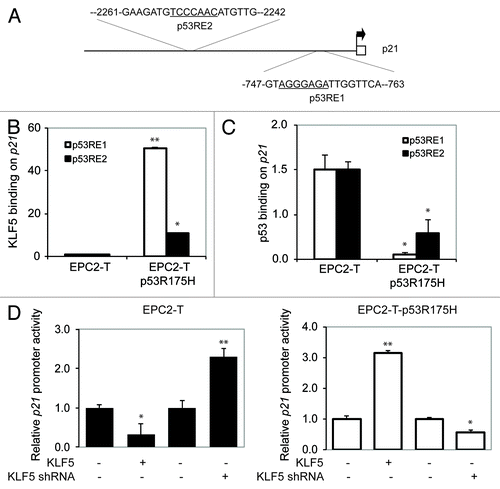
The 5′ regulatory region of p21Waf1/Cip1 contains two putative KLF5-binding sites overlapping two well-defined p53 response elements (p53RE1 and P53RE2)Citation32,Citation33 on p21Waf1/Cip1 (). We hypothesized that KLF5 might therefore bind to p21Waf1/Cip1 in these regions, and that this binding was dependent upon p53 status. In fact, by chromatin immunopreciptation (ChIP), KLF5 bound strongly to p21Waf1/Cip1 at both p53 response elements when p53 was mutant, but not with wild-type p53 (); predictably, p53 binding to the p53 response elements decreased with p53R175H (). This reciprocal binding relationship was similar to that seen previously for Notch1,Citation20 and interestingly, the level of increased KLF5 binding at each response element on p21Waf1/Cip1 was mirrored by the decreases in p53 binding at each site. KLF5 also transcriptionally repressed the p21Waf1/Cip1 promoter when p53 was wild-type but activated the p21Waf1/Cip1 promoter both with p53R175H in EPC2-T cells () and in TE-7 ESCC cells, which lack p53 expressionCitation25 (Fig. S3B).
Figure 3. KLF5 directly binds and transactivates p21Waf1/Cip1 in the presence of p53R175H. (A) The 5′ upstream regulatory region of p21Waf1/Cip1 contains two putative KLF5 binding sites (underlined) within two p53 response elements (p53RE1 and p53RE2). (B) Quantitative ChIP revealed that KLF5 binding on p21Waf1/Cip1 was markedly increased in cells with p53R175H as compared with cells with wild-type p53. (C) A reciprocal pattern was seen for p53 binding to p21Waf1/Cip1. (D) Luciferase reporter assays revealed that KLF5 repressed p21Waf1/Cip1 in EPC2-T cells but was a transcriptional activator of p21Waf1/Cip1 in EPC2-T-p53R175H cells. *p < 0.05, ** p < 0.01
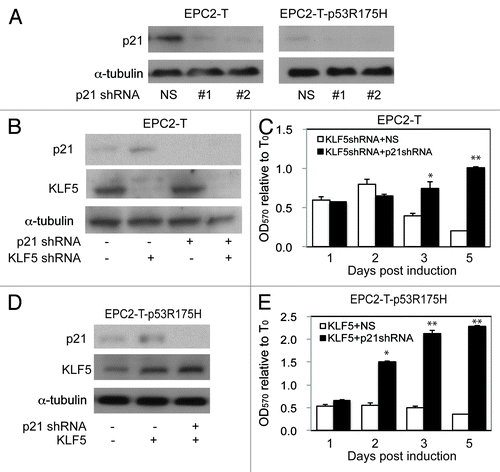
To determine whether p21Waf1/Cip1 is essential for the p53-dependent effects of KLF5 on cell proliferation, we knocked down p21Waf1/Cip1 () in EPC2-T and EPC2-T-p53R175H cells with or without KLF5 induction or KLF5 knockdown. When KLF5 was suppressed in EPC2-T cells with wild-type p53, a condition that yielded increased p21Waf1/Cip1 () and reduced cell proliferation, p21Waf1/Cip1 knockdown blocked the inhibitory effects of KLF5 knockdown on cell proliferation (). Similarly, in EPC2-T-p53R175H cells, where KLF5 induction activated p21Waf1/Cip1 () and decreased cell proliferation, suppression of p21Waf1/Cip1 prevented the growth-inhibitory effect of KLF5 in this context (). In other contexts, where KLF5 activation of p21Waf1/Cip1 is minimal, the effect of p21Waf1/Cip1 knockdown on cell proliferation was also minimal (Fig. S3C). Thus, KLF5 is pro-proliferative or growth-inhibitory depending upon p53 status, and these context-dependent effects of KLF5 are mediated by and require p21Waf1/Cip1.
Figure 4. p21Waf1/Cip1 mediates the p53-dependent effects of KLF5 on cell proliferation. (A) By western blot, p21Waf1/Cip1 shRNA #1 and #2 successfully decreased p21Waf1/Cip1, compared with non-silencing (NS) control. (B and C) In EPC2-T cells with p21Waf1/Cip1 and/or KLF5 knockdown (B), p21Waf1/Cip1 suppression prevented the growth-inhibitory effect of KLF5 knockdown (C). (D and E) EPC2-T-p53R175H cells with KLF5 induction and/or p21Waf1/Cip1 suppression (D) demonstrated that p21Waf1/Cip1 knockdown prevented the growth-inhibitory effect of KLF5 expression in these cells. *p < 0.05, ** p < 0.01
Discussion
Here we demonstrate that pro-proliferative KLF5 becomes anti-proliferative in the presence of p53R175H mutation, one of the most common genetic events in esophageal and other epithelial cancers,Citation26,Citation27 via transcriptional activation of p21Waf1/Cip1. Certainly, the context-dependent effects of KLF5 on cell proliferation may involve factors other than p21Waf1/Cip1, and p15 and p27, for example, are upregulated at specific stages following KLF5 suppression in p53 mutant cells; nonetheless, it is clear that p21Waf1/Cip1 is crucial for these effects, as suppression of p21Waf1/Cip1 alone abrogates the growth-inhibitory effects of KLF5 with p53R175H. Posttranslational modification of KLF5 by TGFβ may also be important in defining the effects of KLF5 on proliferation in some contexts,Citation34 and p53 has been linked to TGFβ signaling.Citation35 In our model, the effects of KLF5 on cell proliferation are cell-autonomous and occur in the absence of any exogenous stimuli, while the use of exogenous TGFβ in a spontaneously immortalized, aneuploid cell line (HaCaT) may limit the relevance of these findings to normal homeostasis. Notably, the effects of KLF5 knockdown on proliferation and p21Waf1/Cip1 in EPC2-T-p53R175H cells occur before the onset of cellular transformation.Citation20
In response to cellular stress, p53 is activated, triggering either apoptosis or, via induction of p21Waf1/Cip1, growth arrest.Citation36-Citation38 However, when p53 cannot activate p21Waf1/Cip1, as in the presence of the p53R175H mutation,Citation39 KLF5 then assumes a key role in cell cycle control and growth arrest. Interestingly, KLF5 is upregulated in esophageal squamous cell dysplasia, a precursor lesion, but lost in esophageal squamous cell cancer.Citation20 In human cancers, it is likely that multiple genetic and/or epigenetic events drive tumor progression, and we believe that upregulation of KLF5 may represent an attempt by the cell to maintain control of cellular proliferation. Since p53 function is typically lost early in esophageal squamous cell carcinogenesis,Citation21,Citation40,Citation41 KLF5 regulation of cell cycle control via p21Waf1/Cip1 is likely critical in dysplastic tissue. When KLF5 is lost, cells transform,Citation20 and proliferation is unrestrained. Thus, KLF5 can promote proliferationCitation13,Citation14 while also providing a critical brake on the system. As such, it would be interesting to examine the effects of cellular stressors, such as irradiation or chemotherapeutics, on the coordinate functions of KLF5 and p53, both in growth arrest and apoptosis, although KLF5 effects on apoptosis may be independent of p53.Citation42 Nonetheless, our findings could help explain the selective proliferative advantages of p53 mutant cells following UV irradiation,Citation43 if KLF5 is also silenced in this context.
Of note, while KLF5 induction repressed the exogenous p21Waf1/Cip1 reporter in EPC2-T cells (), it did not decrease either p21Waf1/Cip1 mRNA levels () or cell proliferation (). Thus, the lack of change in p21Waf1/Cip1 expression in response to KLF5 induction may be more representative of the physiologic state. While KLF5 is one of a group of KLFs that typically function as transcriptional activators, the functionality of each KLF can be dictated through its association with specific co-activators or co-repressors.Citation44 These co-regulators include the histone acetyltransferases p300 and PCAF and the histone deacetylases HDAC1 and HDAC2, all of which can control access of KLFs to promoter DNA.Citation45,Citation46 Additionally, KLF5 and also p53 can be targeted directly by some of these factors, leading to posttranslational modifications which affect function.Citation47-Citation50 Interestingly, PCAF can also alter the ability of p53 to transcriptionally activate p21Waf1/Cip1 independent of DNA binding.Citation47
Notably, while some epithelial cells may undergo EMT in response to p53 loss alone,Citation51,Citation52 keratinocytes require loss of both KLF5 and p53 for EMT.Citation20 Our findings here are consistent with prior reports of a tumor-suppressive role for KLF5 in squamous epitheliaCitation18,Citation20 and other tissues.Citation53,Citation54 Yet other studies have suggested that KLF5 may function as an oncogene, particularly in Ras-mediated transformation.Citation17,Citation55 One way to reconcile these findings is that KLF5 may be important for the early, dysplastic stages of carcinogenesis, such as esophageal squamous cell dysplasia and intestinal adenoma formation, both of which are linked to KLF5 expression.Citation20,Citation56 Alternatively, KLF5 function may depend upon tissue context, and other tissue-specific factors may be involved. In any case, the coordinate transcriptional regulation of p21Waf1/Cip1 by KLF5 and p53 provides a novel and potentially important mechanism for growth control during carcinogenesis, particularly in tumors characterized by p53 mutation or loss. Overall, we conclude that KLF5, which is pro-proliferative in normal epithelial homeostasis, provides a critical brake on an aberrant cell cycle, with important tumor-suppressive functions in esophageal squamous cell and potentially other epithelial cancers.
Materials and Methods
Cell culture
Primary human esophageal keratinocytes transduced with human telomerase reverse transcriptase (EPC2-T) and the oncogenic mutant p53R175H (EPC2-T-p53R175H) were as previously describedCitation20,Citation23,Citation57 and were cultured in keratinocyte serum-free medium (K-SFM, Life Technologies) supplemented with 40 µg/ml bovine pituitary extract, 1.0 ng/ml epidermal growth factor (EGF), 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C and 5% CO2. TE7 esophageal squamous cell cancer cellsCitation58 were grown in modified Eagle’s medium (DMEM)/Ham’s F12 medium with 10% FBS (Sigma-Aldrich).
Viral constructs and infection
Full-length KLF5 cDNA was subcloned into the pRevTre retroviral vector (Clontech). pRevTre-KLF5, control pRev-Tre (EV) and pTreTet-On were transfected into Phoenix-Ampho cells with Lipofectamine 2000 (Life Technologies) following the manufacturer’s instructions. Virus-containing supernatant was harvested from 48–72 h after transfection and filtered with 0.45-µm MicroFunnel filters (Pall Life Sciences). Keratinocytes were infected with viral supernatants at a 1:6 dilution with K-SFM and selected with 300 µg/ml G418 and 1 µg/ml hygromycin beginning 24 h after infection and continued for 14 d. shRNA directed against either KLF5, as previously described,Citation20 or p21Waf1/Cip1 was subcloned into the pTripz lentiviral vector (Thermo Scientific Open Biosystems). pTripz-KLF5shRNA, pTripz-p21shRNA and non-silencing control pTripz-NS shRNA (Thermo Scientific, RHS4743) were packaged by co-transfection with the lentiviral packaging plasmids pCMV-dR8.74 and pMD2.VSVG (Addgene plasmids #22036 and #12259). Infected cells were selected with 1 µg/ml puromycin for 14 d.
Quantitative real-time PCR
Total RNA was isolated with the RNeasy micro kit (Qiagen), and cDNA was synthesized with the High Capacity cDNA Reverse Transcription Kit (Life Technologies). Quantitative real-time PCR was performed in triplicate on three samples for each experimental condition on a StepOne Plus Real-Time PCR System (Life Technologies) using Power SYBR Green Master Mix (Life Technologies). TATA-box-binding protein (TBP) served as an internal control. Primer sequences are listed in Table S1.
Western blotting
For each sample, 30 µg of total protein was separated on a Nupage 4% to 12% Bis-Tris acrylamide gel (Life Technologies) and transferred onto polyvinylidene difluroide membrane (Millipore). Membrane was probed with 1:5,000 rabbit anti-KLF5 antibodyCitation18 or 1:500 mouse anti-p21 antibody (Santa Cruz Biotechnology) in TBST with 5% non-fat milk followed by secondary anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase (GE Healthcare Life Sciences) and developed with Immobilon Western Chemiluminescent HRP Substrate (Millipore). Mouse anti-α-tubulin (Sigma-Aldrich) at 1:5,000 served as an internal control.
Flow cytometry
Cells in monolayer were dissociated with 0.05% trypsin (Life Technologies) and neutralized with soybean trypsin inhibitor (Sigma-Aldrich). After fixation with 4% paraformaldehyde in PBS, cells were stained with 50 µg/ml propidium iodide (Life Technologies) in PBST buffer and analyzed with CellQuest Pro software (BD Biosciences) on a BD Biosciences FACSCalibur flow cytometer. Flow cytometry studies were performed in triplicate.
ChIP assays
ChIP assays were performed with the ChIP assay kit (Millipore) as previously described.Citation13 In brief, cells were cross-linked with 1% formaldehyde for 10 min at room temperature, lysed with SDS buffer, and DNA was sheared by sonication. Samples were diluted and pre-cleared with protein A-agarose/salmon sperm for 30 min at 4°C. DNA-protein complexes were incubated with 1:5,000 rabbit anti-KLF5Citation18 or 1:1,000 mouse anti-p53 (Vector Laboratories) antibodies at 4°C overnight and precipitated with protein A-agarose for 1 h. DNA was purified with the Qiaquick PCR purification kit (Qiagen), and quantitative PCR on the p21Waf1/Cip1 promoter was performed with Power SYBR Green Master Mix (Life Technologies) using primers listed in Table S2. Putative-binding sites were identified using the Transcription Element Search System (TESS).Citation59
Luciferase reporter assays
For luciferase reporter assays, we utilized a plasmid containing 2.4 kb of the p21Waf1/Cip1 promoter upstream of the luciferase coding sequence (Addgene plasmid #16451).Citation31,Citation33 EPC2-T or EPC2-T-p53R175H cells, transduced with the viruses outlined above, were transfected using Fugene 6 reagent (Roche Applied Science). The cells were lysed with cell lysis buffer (BD Biosciences), and luciferase activity was measured with the Dual-Luciferase Reporter assay system (Promega) on a Microtiter Plate Luminometer (Dynex Technologies). Data were normalized to Renilla activity and expressed as relative luciferase activity.
TUNEL assays
TUNEL assays were performed using the in situ Cell Death Detection Kit (Roche Applied Science), and the numbers of positive cells were determined by counting in five 200x fields.
Statistical analyses
Results were expressed as mean ± SEM, with statistical significance of differences between experimental conditions established at 95%. Student’s t-test was performed using the Analysis ToolPak for Excel (Microsoft).
| Abbreviations: | ||
| KLF | = | Krüppel-like factor |
| KLF5 | = | Krüppel-like factor 5 |
| ESCC | = | esophageal squamous cell cancer |
| ChIP | = | chromatin immunopreciptation |
Additional material
Download Zip (751.8 KB)Acknowledgments
We thank Dr. Anil Rustgi (University of Pennsylvania) for the EPC2 cell lines. This work was supported by NIH NIDDK R01 DK080031 and DK080031–02S1 to J.P.K. and by the University of Pennsylvania Center for Molecular Studies in Digestive and Liver Diseases (NIH NIDDK P30 DK050306) through the Molecular Pathology and Imaging Core, the Cell Culture Core and the Molecular Biology/Gene Expression Core, and by NIH NCI P01 CA098101 (“Mechanisms of Esophageal Carcinogenesis”).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Richmond CA, Breault DT. Regulation of gene expression in the intestinal epithelium. Prog Mol Biol Transl Sci 2010; 96:207 - 29; http://dx.doi.org/10.1016/B978-0-12-381280-3.00009-9; PMID: 21075346
- Radtke F, Clevers H, Riccio O. From gut homeostasis to cancer. Curr Mol Med 2006; 6:275 - 89; http://dx.doi.org/10.2174/156652406776894527; PMID: 16712475
- Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science 2005; 307:1904 - 9; http://dx.doi.org/10.1126/science.1104815; PMID: 15790842
- Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol 2004; 20:695 - 723; http://dx.doi.org/10.1146/annurev.cellbio.20.010403.092805; PMID: 15473857
- Tetreault MP, Wang ML, Yang Y, Travis J, Yu QC, Klein-Szanto AJ, et al. Klf4 overexpression activates epithelial cytokines and inflammation-mediated esophageal squamous cell cancer in mice. Gastroenterology 2010; 139:2124 - 34, e9; PMID: 20816834
- Tetreault MP, Yang Y, Travis J, Yu QC, Klein-Szanto A, Tobias JW, et al. Esophageal squamous cell dysplasia and delayed differentiation with deletion of krüppel-like factor 4 in murine esophagus. Gastroenterology 2010; 139:171 - 81, e9; PMID: 20347813
- Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet 1999; 22:356 - 60; http://dx.doi.org/10.1038/11926; PMID: 10431239
- Foster KW, Liu Z, Nail CD, Li X, Fitzgerald TJ, Bailey SK, et al. Induction of KLF4 in basal keratinocytes blocks the proliferation-differentiation switch and initiates squamous epithelial dysplasia. Oncogene 2005; 24:1491 - 500; http://dx.doi.org/10.1038/sj.onc.1208307; PMID: 15674344
- Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer 2006; 6:11 - 23; http://dx.doi.org/10.1038/nrc1780; PMID: 16372018
- Vooijs M, Liu Z, Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology 2011; 141:448 - 59; http://dx.doi.org/10.1053/j.gastro.2011.06.003; PMID: 21689653
- Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene 2008; 27:5124 - 31; http://dx.doi.org/10.1038/onc.2008.226; PMID: 18758481
- Dotto GP. Notch tumor suppressor function. Oncogene 2008; 27:5115 - 23; http://dx.doi.org/10.1038/onc.2008.225; PMID: 18758480
- Yang Y, Goldstein BG, Nakagawa H, Katz JP. Krüppel-like factor 5 activates MEK/ERK signaling via EGFR in primary squamous epithelial cells. FASEB J 2007; 21:543 - 50; http://dx.doi.org/10.1096/fj.06-6694com; PMID: 17158781
- Goldstein BG, Chao HH, Yang Y, Yermolina YA, Tobias JW, Katz JP. Overexpression of Kruppel-like factor 5 in esophageal epithelia in vivo leads to increased proliferation in basal but not suprabasal cells. Am J Physiol Gastrointest Liver Physiol 2007; 292:G1784 - 92; http://dx.doi.org/10.1152/ajpgi.00541.2006; PMID: 17395897
- McConnell BB, Ghaleb AM, Nandan MO, Yang VW. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays 2007; 29:549 - 57; http://dx.doi.org/10.1002/bies.20581; PMID: 17508399
- Nandan MO, Chanchevalap S, Dalton WB, Yang VW. Krüppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett 2005; 579:4757 - 62; http://dx.doi.org/10.1016/j.febslet.2005.07.053; PMID: 16102754
- Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW. Krüppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene 2004; 23:3404 - 13; http://dx.doi.org/10.1038/sj.onc.1207397; PMID: 15077182
- Yang Y, Goldstein BG, Chao HH, Katz JP. KLF4 and KLF5 regulate proliferation, apoptosis and invasion in esophageal cancer cells. Cancer Biol Ther 2005; 4:1216 - 21; http://dx.doi.org/10.4161/cbt.4.11.2090; PMID: 16357509
- Bateman NW, Tan D, Pestell RG, Black JD, Black AR. Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem 2004; 279:12093 - 101; http://dx.doi.org/10.1074/jbc.M311532200; PMID: 14726538
- Yang Y, Nakagawa H, Tetreault MP, Billig J, Victor N, Goyal A, et al. Loss of transcription factor KLF5 in the context of p53 ablation drives invasive progression of human squamous cell cancer. Cancer Res 2011; 71:6475 - 84; http://dx.doi.org/10.1158/0008-5472.CAN-11-1702; PMID: 21868761
- Gao H, Wang LD, Zhou Q, Hong JY, Huang TY, Yang CS. p53 tumor suppressor gene mutation in early esophageal precancerous lesions and carcinoma among high-risk populations in Henan, China. Cancer Res 1994; 54:4342 - 6; PMID: 8044781
- Hollstein MC, Metcalf RA, Welsh JA, Montesano R, Harris CC. Frequent mutation of the p53 gene in human esophageal cancer. Proc Natl Acad Sci USA 1990; 87:9958 - 61; http://dx.doi.org/10.1073/pnas.87.24.9958; PMID: 2263646
- Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res 2003; 1:729 - 38; PMID: 12939398
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 1988; 106:761 - 71; http://dx.doi.org/10.1083/jcb.106.3.761; PMID: 2450098
- Barnas C, Martel-Planche G, Furukawa Y, Hollstein M, Montesano R, Hainaut P. Inactivation of the p53 protein in cell lines derived from human esophageal cancers. Int J Cancer 1997; 71:79 - 87; http://dx.doi.org/10.1002/(SICI)1097-0215(19970328)71:1<79::AID-IJC14>3.0.CO;2-4; PMID: 9096669
- Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 2007; 26:2157 - 65; http://dx.doi.org/10.1038/sj.onc.1210302; PMID: 17401424
- Soussi T, Asselain B, Hamroun D, Kato S, Ishioka C, Claustres M, et al. Meta-analysis of the p53 mutation database for mutant p53 biological activity reveals a methodologic bias in mutation detection. Clin Cancer Res 2006; 12:62 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-05-0413; PMID: 16397025
- Dulić V, Stein GH, Far DF, Reed SI. Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M-phase transition. Mol Cell Biol 1998; 18:546 - 57; PMID: 9418901
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 501; http://dx.doi.org/10.1126/science.282.5393.1497; PMID: 9822382
- Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187 - 90; PMID: 7585571
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75:817 - 25; http://dx.doi.org/10.1016/0092-8674(93)90500-P; PMID: 8242752
- Wang B, Xiao Z, Ren EC. Redefining the p53 response element. Proc Natl Acad Sci USA 2009; 106:14373 - 8; http://dx.doi.org/10.1073/pnas.0903284106; PMID: 19597154
- el-Deiry WS, Tokino T, Waldman T, Oliner JD, Velculescu VE, Burrell M, et al. Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res 1995; 55:2910 - 9; PMID: 7796420
- Guo P, Dong XY, Zhang X, Zhao KW, Sun X, Li Q, et al. Pro-proliferative factor KLF5 becomes anti-proliferative in epithelial homeostasis upon signaling-mediated modification. J Biol Chem 2009; 284:6071 - 8; http://dx.doi.org/10.1074/jbc.M806270200; PMID: 19056724
- Dupont S, Zacchigna L, Adorno M, Soligo S, Volpin D, Piccolo S, et al. Convergence of p53 and TGF-beta signaling networks. Cancer Lett 2004; 213:129 - 38; http://dx.doi.org/10.1016/j.canlet.2004.06.008; PMID: 15327827
- Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2010; 2:a000893; http://dx.doi.org/10.1101/cshperspect.a000893; PMID: 20463001
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402 - 12; http://dx.doi.org/10.1038/nrm2395; PMID: 18431400
- Hill R, Madureira PA, Waisman DM, Lee PW. DNA-PKCS binding to p53 on the p21WAF1/CIP1 promoter blocks transcription resulting in cell death. Oncotarget 2011; 2:1094 - 108; PMID: 22190353
- Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2010; 2:a001107; http://dx.doi.org/10.1101/cshperspect.a001107; PMID: 20182618
- Shi ST, Yang GY, Wang LD, Xue Z, Feng B, Ding W, et al. Role of p53 gene mutations in human esophageal carcinogenesis: results from immunohistochemical and mutation analyses of carcinomas and nearby non-cancerous lesions. Carcinogenesis 1999; 20:591 - 7; http://dx.doi.org/10.1093/carcin/20.4.591; PMID: 10223186
- Yasuda M, Kuwano H, Watanabe M, Toh Y, Ohno S, Sugimachi K. p53 expression in squamous dysplasia associated with carcinoma of the oesophagus: evidence for field carcinogenesis. Br J Cancer 2000; 83:1033 - 8; http://dx.doi.org/10.1054/bjoc.2000.1443; PMID: 10993651
- Zhao Y, Hamza MS, Leong HS, Lim CB, Pan YF, Cheung E, et al. Kruppel-like factor 5 modulates p53-independent apoptosis through Pim1 survival kinase in cancer cells. Oncogene 2008; 27:1 - 8; http://dx.doi.org/10.1038/sj.onc.1210625; PMID: 17603560
- Benjamin CL, Ananthaswamy HN. p53 and the pathogenesis of skin cancer. Toxicol Appl Pharmacol 2007; 224:241 - 8; http://dx.doi.org/10.1016/j.taap.2006.12.006; PMID: 17270229
- McConnell BB, Yang VW. Mammalian Krüppel-like factors in health and diseases. Physiol Rev 2010; 90:1337 - 81; http://dx.doi.org/10.1152/physrev.00058.2009; PMID: 20959618
- Geiman DE, Ton-That H, Johnson JM, Yang VW. Transactivation and growth suppression by the gut-enriched Krüppel-like factor (Krüppel-like factor 4) are dependent on acidic amino acid residues and protein-protein interaction. Nucleic Acids Res 2000; 28:1106 - 13; http://dx.doi.org/10.1093/nar/28.5.1106; PMID: 10666450
- Zhang JS, Moncrieffe MC, Kaczynski J, Ellenrieder V, Prendergast FG, Urrutia R. A conserved α-helical motif mediates the interaction of Sp1-like transcriptional repressors with the corepressor mSin3A. Mol Cell Biol 2001; 21:5041 - 9; http://dx.doi.org/10.1128/MCB.21.15.5041-5049.2001; PMID: 11438660
- Love IM, Sekaric P, Shi D, Grossman SR, Androphy EJ. The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 2012; 11:2458 - 66; http://dx.doi.org/10.4161/cc.20864; PMID: 22713239
- Matsumura T, Suzuki T, Aizawa K, Munemasa Y, Muto S, Horikoshi M, et al. The deacetylase HDAC1 negatively regulates the cardiovascular transcription factor Krüppel-like factor 5 through direct interaction. J Biol Chem 2005; 280:12123 - 9; http://dx.doi.org/10.1074/jbc.M410578200; PMID: 15668237
- Miyamoto S, Suzuki T, Muto S, Aizawa K, Kimura A, Mizuno Y, et al. Positive and negative regulation of the cardiovascular transcription factor KLF5 by p300 and the oncogenic regulator SET through interaction and acetylation on the DNA-binding domain. Mol Cell Biol 2003; 23:8528 - 41; http://dx.doi.org/10.1128/MCB.23.23.8528-8541.2003; PMID: 14612398
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997; 90:595 - 606; http://dx.doi.org/10.1016/S0092-8674(00)80521-8; PMID: 9288740
- Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 2011; 13:317 - 23; http://dx.doi.org/10.1038/ncb2173; PMID: 21336307
- Pinho AV, Rooman I, Real FX. p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle 2011; 10:1312 - 21; http://dx.doi.org/10.4161/cc.10.8.15363; PMID: 21490434
- Chen C, Bhalala HV, Qiao H, Dong JT. A possible tumor suppressor role of the KLF5 transcription factor in human breast cancer. Oncogene 2002; 21:6567 - 72; http://dx.doi.org/10.1038/sj.onc.1205817; PMID: 12242654
- Chen C, Bhalala HV, Vessella RL, Dong JT. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate 2003; 55:81 - 8; http://dx.doi.org/10.1002/pros.10205; PMID: 12661032
- Nandan MO, McConnell BB, Ghaleb AM, Bialkowska AB, Sheng H, Shao J, et al. Krüppel-like factor 5 mediates cellular transformation during oncogenic KRAS-induced intestinal tumorigenesis. Gastroenterology 2008; 134:120 - 30; http://dx.doi.org/10.1053/j.gastro.2007.10.023; PMID: 18054006
- McConnell BB, Bialkowska AB, Nandan MO, Ghaleb AM, Gordon FJ, Yang VW. Haploinsufficiency of Krüppel-like factor 5 rescues the tumor-initiating effect of the Apc(Min) mutation in the intestine. Cancer Res 2009; 69:4125 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-08-4402; PMID: 19435907
- Kim SH, Nakagawa H, Navaraj A, Naomoto Y, Klein-Szanto AJ, Rustgi AK, et al. Tumorigenic conversion of primary human esophageal epithelial cells using oncogene combinations in the absence of exogenous Ras. Cancer Res 2006; 66:10415 - 24; http://dx.doi.org/10.1158/0008-5472.CAN-06-2104; PMID: 17079462
- Boonstra JJ, van der Velden AW, Beerens EC, van Marion R, Morita-Fujimura Y, Matsui Y, et al. Mistaken identity of widely used esophageal adenocarcinoma cell line TE-7. Cancer Res 2007; 67:7996 - 8001; http://dx.doi.org/10.1158/0008-5472.CAN-07-2064; PMID: 17804709
- Schug J. Using TESS to predict transcription factor binding sites in DNA sequence. Curr Protoc Bioinformatics 2008; Chapter 2:Unit 2 6.