Abstract
Erlotinib was originally developed as an epidermal growth factor receptor (EGFR)-specific inhibitor for the treatment of solid malignancies, yet also exerts significant EGFR-independent antileukemic effects in vitro and in vivo. The molecular mechanisms underlying the clinical antileukemic activity of erlotinib as a standalone agent have not yet been precisely elucidated. Conversely, in preclinical settings, erlotinib has been shown to inhibit the constitutive activation of SRC kinases and mTOR, as well as to synergize with the DNA methyltransferase inhibitor azacytidine (a reference therapeutic for a subset of leukemia patients) by promoting its intracellular accumulation. Here, we show that both erlotinib and gefitinib (another EGFR inhibitor) inhibit transmembrane transporters of the ATP-binding cassette (ABC) family, including P-glycoprotein (P-gp), multidrug resistance-associated proteins (MRPs) and breast cancer resistance protein (BCRP), also in acute myeloid leukemia (AML) cells that do not overexpress these pumps. Thus, inhibition of drug efflux by erlotinib and gefitinib selectively exacerbated (in a synergistic or additive fashion) the cytotoxic response of KG-1 cells to chemotherapeutic agents that are normally extruded by ABC transporters (e.g., doxorubicin and etoposide). Erlotinib limited drug export via ABC transporters by multiple mechanisms, including the downregulation of surface-exposed pumps and the modulation of their ATPase activity. The effects of erlotinib on drug efflux and its chemosensitization profile persisted in patient-derived CD34+ cells, suggesting that erlotinib might be particularly efficient in antagonizing leukemic (stem cell) subpopulations, irrespective of whether they exhibit or not increased drug efflux via ABC transporters.
Introduction
Primary or secondary treatment failure in acute myeloid leukemia (AML) is frequently due to the persistence of leukemic progenitor cells that are inherently resistant against classical chemotherapeutic agents.Citation1,Citation2 This cell population is often characterized by an increased efflux of chemotherapeutics via transmembrane pumps of the ATP-binding cassette (ABC) family, including ABCB1, also known as P-glycoprotein (P-gp), ABCC1, also known as multidrug resistance-associated protein 1 (MRP1), and ABCG2, also known as breast cancer resistance protein (BCRP).Citation3,Citation4 In line with this notion, increased expression and functionality of ABC transporters on the surface of AML cells have been correlated with poor prognosis due to chemoresistance.Citation5,Citation6 Notably, ABC transporters are frequently overexpressed in AML cases that exhibit additional risk factors, such as an unfavorable karyotype or derivation from a myelodysplastic syndrome (MDS).Citation7 In addition, recent clinical studies indicate that the activity of ABC transporters constitutes an independent prognostic factor even upon consideration of NPM and FLT3 mutational status.Citation8
Based on these premises, it has been hypothesized that the inhibition of ABC transporters might restore sensitivity to chemotherapy in high-risk AML patients and allow for leukemia eradication.Citation9 Nevertheless, most clinical studies performed so far have failed to demonstrate a significant increase in survival when ABC pump (in particular P-gp) inhibitors were combined with classical chemotherapeutic regimens,Citation5 possibly due to the elevated degree of redundancy of ABC transporters.Citation10
The small molecules erlotinib and gefitinib were originally developed to inhibit the kinase activity of the epidermal growth factor receptor (EGFR) in solid neoplasms,Citation11,Citation12 yet they exhibit in vitro and in vivo efficacy against MDS and AML, even though blasts generally do not express EGFR.Citation13-Citation15 In particular, two case-report studies have demonstrated that erlotinib alone can induce durable and complete remissions in AML patients.Citation16,Citation17 Two studies (including one from our group) that rigorously assess the tolerance and therapeutic potential of erlotinib in MDS and AML patients are currently registered at www.clinicaltrials.gov (NCT00977548, NCT01085838), and formal evidence for an antileukemic efficacy of erlotinib, at least in a subset of patients, is emerging.Citation18 However, the molecular mechanisms whereby erlotinib as a standalone agent exerts clinical antileukemic activity have not yet been precisely elucidated.
Recently, we have demonstrated that erlotinib synergizes with the DNA methyltransferase inhibitor 5-azacytidine (azacytidine), but not with its functional analog decitabine, in the killing of AML-derived cell lines and patient blasts in vitro.Citation19 Such a synergistic antileukemic effect stemmed from a pharmacokinetic mechanism involving an increased intracellular accumulation of azacytidine.Citation19 Of note, EGFR-targeting agents have previously been shown to interfere with the activity of P-gpCitation20-Citation22 and BCRPCitation21,Citation22 in multidrug-resistant (MDR) leukemic cells. However, we were unable to find any study addressing the possibility that erlotinib and gefitinib might influence the accumulation (and hence the cytotoxicity) of antileukemic drugs in cells that do not overexpress ABC transporters.
Therefore, we decided to determine if and how erlotinib and gefitinib antagonize drug extrusion via ABC pumps in KG-1 AML cells, which are known to express limited amounts of P-gp,Citation23 BCRPCitation24 and MDR-1.Citation25 Here, we demonstrate that EGFR-targeting chemicals inhibit the efflux of specific chemotherapeutics from leukemic cells even when these do not overexpress ABC transporters, hence exacerbating drug cytotoxicity in a synergistic or additive fashion. Such a chemosensitization effect persisted in patient-derived blasts, suggesting that the combination of erlotinib and conventional regimens may provide therapeutic benefits to MDS or AML patients.
Results
EGFR-targeting agents enhance the chemosensitivity of AML cells
In order to evaluate whether erlotinib and gefitinib might chemosensitize AML-derived cell lines to conventional chemotherapeutics, we monitored the death of KG-1 cells upon exposure to drugs that are commonly used in the treatment of AML, notably cytarabine, doxorubicin or etoposide,Citation26 alone or in combination with EGFR inhibitors. In line with previous reports,Citation14,Citation15 erlotinib and gefitinib per se induced a moderate increase in the percentage of KG-1 cells exhibiting mitochondrial transmembrane potential (Δψm) dissipation (a sign of imminent cell death) and plasma membrane breakdown (), as assessed by cytofluorometry upon co-staining with the Δψm-sensitive cationic fluorochrome DiOC6(3) and the vital dye propidium iodide (PI).Citation27-Citation29 While failing to affect the response of KG-1 cells to cytarabine, erlotinib and gefitinib potentiated etoposide-induced (and to a lesser degree, doxorubicin-induced) cell death (). This chemosensitizing effect was apparent as early as 24 h after the administration of drugs, increased over time and was most pronounced 72 h after stimulation (). To delineate this phenomenon more precisely, we administered KG-1 cells with 1 μM etoposide combined with a broader range of erlotinib and gefitinib concentrations (1, 5 and 10 μM). We found that, while EGFR-targeting agents at doses ≤ 1 μM fail to sensitize KG-1 cells to etoposide cytotoxicity, chemosensitization starts at 5 μM and is well-pronounced at 10 μM (). We then analyzed the interaction between EGFR inhibitors and etoposide by employing a colorimetric test of cell viability/proliferation and by calculating the combination index (CI) following Harbron’s method.Citation30 The combination of etoposide and erlotinib (), but not gefitinib (Fig. S1), was associated with a CI < 1 over a wide range of concentrations, demonstrating that these two agents de facto synergize in the killing of KG-1 cells.
Figure 1. Erlotinib and gefitinib increase the sensitivity of KG-1 cells to etoposide. (A–C). KG-1 cells were left untreated or incubated with the indicated concentration of erlotinib (ERLO, 10 μM where not specified), gefitinib (GEFI, 10 μM where not specified), etoposide (VP16, 1 μM where not specified), 0.1 μM cytarabine (ARAC), 0.1 μM doxorubicin (DOXO), alone or in combination, for the indicated time (72 h where not specified), followed by DiOC6(3)/PI co-staining for the determination of apoptosis-related parameters. In (A), representative dot plots are reported, and the percentage of cells exhibiting mitochondrial transmembrane potential dissipation [PI- DiOC6(3)low] or the breakdown of plasma membrane (PI+) is indicated. (B and C) depict quantitative data (means ± SEM; n = 3). **p < 0.01, ***p < 0.001 (ANOVA plus Bonferroni’s post-hoc test), as compared with DMSO-treated cells; #p < 0.05, ###p < 0.001 (ANOVA plus Bonferroni’s post-hoc test), as compared with cells treated with EGFR inhibitors alone; ††p < 0.01, †††p < 0.001 (ANOVA plus Bonferroni’s post-hoc test), as compared with cells treated with VP16 alone; ¤p < 0.05 (ANOVA plus Bonferroni’s post-hoc test), as compared with cells treated with DOXO alone. (D). KG-1 cells were left untreated or incubated with the indicated concentrations of ERLO alone or combined with 0.1–2 μM VP16 for 48 h, followed by colorimetric determinations of cell viability/proliferation. Combination index (CI) values as calculated—according to the Harbron’s method—for the indicated concentrations of ERLO as well as for the global data set are reported (means ± 95% confidence interval).
![Figure 1. Erlotinib and gefitinib increase the sensitivity of KG-1 cells to etoposide. (A–C). KG-1 cells were left untreated or incubated with the indicated concentration of erlotinib (ERLO, 10 μM where not specified), gefitinib (GEFI, 10 μM where not specified), etoposide (VP16, 1 μM where not specified), 0.1 μM cytarabine (ARAC), 0.1 μM doxorubicin (DOXO), alone or in combination, for the indicated time (72 h where not specified), followed by DiOC6(3)/PI co-staining for the determination of apoptosis-related parameters. In (A), representative dot plots are reported, and the percentage of cells exhibiting mitochondrial transmembrane potential dissipation [PI- DiOC6(3)low] or the breakdown of plasma membrane (PI+) is indicated. (B and C) depict quantitative data (means ± SEM; n = 3). **p < 0.01, ***p < 0.001 (ANOVA plus Bonferroni’s post-hoc test), as compared with DMSO-treated cells; #p < 0.05, ###p < 0.001 (ANOVA plus Bonferroni’s post-hoc test), as compared with cells treated with EGFR inhibitors alone; ††p < 0.01, †††p < 0.001 (ANOVA plus Bonferroni’s post-hoc test), as compared with cells treated with VP16 alone; ¤p < 0.05 (ANOVA plus Bonferroni’s post-hoc test), as compared with cells treated with DOXO alone. (D). KG-1 cells were left untreated or incubated with the indicated concentrations of ERLO alone or combined with 0.1–2 μM VP16 for 48 h, followed by colorimetric determinations of cell viability/proliferation. Combination index (CI) values as calculated—according to the Harbron’s method—for the indicated concentrations of ERLO as well as for the global data set are reported (means ± 95% confidence interval).](/cms/asset/14e4655d-2928-4f98-8e00-4b127037d662/kccy_a_10922382_f0001.gif)
Erlotinib increases the intracellular level of chemotherapeutic agents by limiting drug efflux via ATP-binding cassette (ABC) transporters
To understand whether the chemosensitizing potential of erlotinib and gefitinib is linked to decreased drug extrusion also in KG-1 cells, which do not overexpress ABC pumps,Citation23-Citation25 we quantified intracellular etoposide levels by high performance liquid chromatography and mass spectrometry (). KG-1 cells co-treated with erlotinib accumulated twice the amount of etoposide than KG-1 cells treated with etoposide alone (). Similarly, the intracellular levels of doxorubicin were increased in the presence of erlotinib (data not shown). To corroborate the finding that erlotinib increases the intracellular level of chemotherapeutic agents exported by ABC transporters, we took advantage of the fact that two clinically employed drugs, doxorubicin and mitoxantrone, are autofluorescent and emit at 575 and 670 nm, respectively. This allows for the straightforward assessment of the intracellular drug levels by cytofluorometry.Citation4 Emission profiles confirmed that EGFR-targeting agents increase the quantity of doxorubicin and mitoxantrone that is retained intracellularly (Fig. 2B–E). In line with cytotoxicity data (), this effect of erlotinib and gefitinib in KG-1 cells was negligible at 1, evident at 5 and well-pronounced at 10 μM.
Figure 2. Erlotinib exacerbates the intracellular retention of etoposide, doxorubicin and mitoxantrone. (A–E). KG-1 cells were left untreated or incubated with the indicated concentration of erlotinib (ERLO, 10 μM where not specified), gefitinib (GEFI, 10 μM where not specified), 0.1 μM doxorubicin (DOXO), 0.3 μM mitoxantrone (MITO), 1 μM etoposide (VP16), 1 μM cyclosporine A (CsA), 0.5 μM KO-143, 10 μM MK-571, 10 μM verapamil (VERA), alone or combined as indicated (4 INH = CsA + KO-143 + MK-571 + VERA) for 1, 12 or 24 h, then subjected to mass spectrometry (A) or cytofluorometry (B–E) for the quantification of intracellular VP16 (A), DOXO (B and C) or MITO (D and E), respectively. In (A), the area under the curve of the VP16 signal (an indicator of the intracellular concentration of VP16) is indicated. (B and D) report representative fluorescence distributions for DOXO and MITO, respectively. In (C and E), quantitative data are reported (fluorescence normalized to that of cells treated with DOXO and MITO only, means ± SEM; n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ns = non-significant (ANOVA plus Dunnett’s test), as compared with cells loaded with DOXO (C) or MITO (E) only. ISO, isotype control.
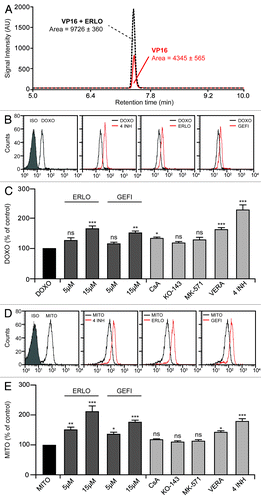
We then sought to identify the specific ABC transporter(s) that is/are inhibited by erlotinib to reduce drug efflux. To this aim, we took advantage of a panel of biochemical inhibitors, namely cyclosporine A (CsA, a P-gp-specific agent), KO-143 (which inhibits efflux via BCRP), MK-571 (an inhibitor of MRPs) and verapamil (a mixed P-gp/BCRP inhibitor).Citation5,Citation31 None of the monospecific inhibitors (i.e., CsA, KO-143 and MK-571) used as a single agent was significantly superior to erlotinib and gefitinib (employed at the optimal concentration of 15 μM) in limiting the efflux of doxorubicin (). Similarly, CsA, KO-143 and MK-571 all failed to significantly increase the intracellular levels of mitoxantrone in conditions in which erlotinib and gefitinib alone efficiently did so (). The mixed P-gp/BCRP inhibitor verapamil was more efficient than CsA, KO-143 and MK-571 in inhibiting the extrusion of both doxorubicin () and mitoxantrone (). Noteworthy, the combined used of CsA, KO-143, MK-571 and verapamil resulted in an approximately 2-fold increase in the intracellular concentration of doxorubicin and mitoxantrone, an effect that was comparable to that of optimal concentrations of erlotinib (). These observations suggest that blocking one single type of ABC transporter cannot increase the intracellular level of its substrate, most likely due to a high degree of functional redundancy.Citation10 The fact that erlotinib and gefitinib per se promoted the intracellular accumulation of doxorubicin and mitoxantrone (in the same conditions in which P-gp, MRP and BCRP inhibitors almost invariably failed to do so) strongly suggests that EGFR inhibitors modulate drug efflux through their simultaneous action on several efflux pumps.
To determine which ABC transporters are inhibited by erlotinib, we employed fluorescent compounds that are preferentially extruded by cells via one specific ABC pump. In particular, we employed 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)] and rhodamine 123 (Rh123) to evaluate the activity of P-gp, calcein and 5-(and-6)-carboxy-2′,7′ diclorofluorescein diacetate (CDCFDA) to follow MRP-mediated efflux, and Hoechst 33342 to monitor the functionality of BCRP.Citation32-Citation34 Incubation of KG-1 cells with erlotinib or gefitinib increased the intracellular accumulation of DiOC2(3) () and Rh123 (data not shown) in a dose-dependent manner, yet this retention effect was not as pronounced as that exerted by CsA (which reportedly blocks P-gp) (). Conversely, erlotinib and gefitinib were capable of increasing the intracellular accumulation of calcein () and CDCFDA (data not shown) with a potency that was comparable to that of the MRP inhibitor MK-571 (). Only a minor fraction (~6%) of KG-1 cells failed to accumulate Hoechst 33342 (). This “side population” is characterized by increased drug efflux and reportedly exhibits other cell stem-like features.Citation4 The dimension of the side population was reduced upon incubation of KG1 cells with verapamil or erlotinib (). Strikingly, the proportion of KG-1a cells, a more undifferentiated variant of the KG-1 cell line,Citation35 that efficiently extruded Hoechst 33342 was close to 80% (). Again, this side population was reduced to < 20% when KG-1a cells were co-incubated with either verapamil or erlotinib (). Immunoblotting assessments revealed that the protein levels of BCRP are significantly higher in KG-1a than in KG-1 cells (), in line with the fact that KG-1a cells contain a much larger side population than KG-1 cells ().
Figure 3. Erlotinib inhibits drug efflux via P-gp, BCRP and MRPs. (A–D). KG-1 cells were loaded with 20 nM DiOC2(3) (A and B) and 10 nM calcein (C and D) alone or together with the indicated concentration of erlotinib (ERLO, 10 μM where not specified), cyclosporine A (CsA, 1 μM where not specified), or 10 μM MK-571 for 2 h, then subjected to cytofluorometry for the determination of DiOC2(3) and calcein fluorescence. Representative profiles and quantitative data (fluorescence normalized to that of cells treated with DiOC2(3) and calcein only, means ± SEM; n = 3) are reported. *p < 0.05, **p < 0.01, ***p < 0.001, ns = non-significant (ANOVA plus Dunnett’s test), as compared with cells loaded with DiOC2(3) (B) or calcein (D) only. (E–G). Alternatively, KG-1 (E and F) and KG-1a (E and G) cells were treated with 1.5 μM Hoechst 33342 (H33342) alone or in combination with 50 μM VERA or 10 μM ERLO for 4 h, followed by the quantification of H33342 fluorescence by cytofluorometry. (E) depicts representative dot plots and the percentage of cells that do not accumulate H33342 (H33342low cells) is indicated. In (F and G), quantitative data are reported (means ± SEM; n = 3). **p < 0.01, ***p < 0.001 (ANOVA plus Dunnett’s test), as compared with cells loaded with H33342 only (F and G). In the inset in (G), untreated KG-1 and KG-1a cells were subjected to immunoblotting for the estimation of breast cancer resistance protein (BCRP) expression levels. The amount of β actin was monitored to ensure equal loading of lanes.
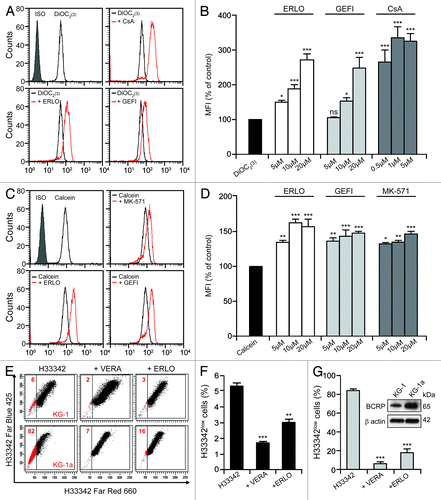
Altogether, these data demonstrate that erlotinib and gefitinib reduce drug efflux by inhibiting more than one single type of ABC transporter.
Erlotinib increases P-gp ATPase activity and reduces P-gp exposure on the surface of KG-1 cells
Next, we investigated if and how erlotinib affects the ATPase activity of P-gp. As a positive control for these determinations, we took advantage of the fact that verapamil is a known substrate of P-gp and an activator of its enzymatic activity.Citation36 Chemiluminescence-based tests confirmed that verapamil can increase the P-gp-mediated consumption of ATP by approximately 3-fold and revealed that erlotinib is even more efficient than verapamil in stimulating the ATPase activity of P-gp (). A similar increase could not be observed with other chemicals that we employed in this study including cytarabine, CsA, doxorubicin and etoposide (both of which actually inhibited the ATPase activity of P-gp) (). Intriguingly, erlotinib stimulated ATP consumption by P-gp with the same efficacy across doses ranging from 5–50 μM (), an activity profile reminiscent of saturatable enzymatic kinetics that did not correlate with the dose-dependent effects mediated by erlotinib on the accumulation of doxorubicin () and of the P-gp substrate DiOC2(3) ().
Figure 4A–C. Erlotinib augments P-gp ATPase activity and inhibits its exposure on the cell surface. (A and B). ATPase activity of recombinant P-gp incubated with the indicated concentration of erlotinib (ERLO, E, 10 μM where not specified), 1 μM cyclosporine A (CsA), 10 μM verapamil (VERA), 0.1 μM cytarabine (ARAC, A), 0.1 μM doxorubicin (DOXO) or 1 μM etoposide (VP16). Results are normalized to basal ATPase activity and reported as means ± SEM (n = 3). *p < 0.05, ***p < 0.001, ns = non-significant (ANOVA plus Dunnett’s test), as compared with basal ATPase activity (A). (C-F). KG-1 cells were kept in control conditions (DMSO) or incubated with the indicated concentration of ERLO (10 μM where not specified), CsA (1 μM where not specified), VERA (10 μM where not specified), 0.1 μM ARAC, 0.1 μM DOXO or 1 μM VP16, alone or combined as indicated, then subjected to cytofluorometry (C–F) or immunoblotting (D) for the quantification of surface-exposed or total P-glycoprotein (P-gp). (C) reports representative profiles of surface-exposed and total P-gp in control conditions.
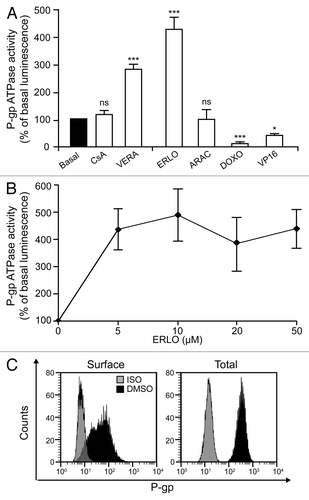
We therefore reasoned that erlotinib might also alter P-gp expression and/or its exposure on the cell surface. To test this hypothesis, we cytofluorometrically quantified P-gp levels on intact and permeabilized KG-1 cells that had previously been exposed to erlotinib or cytarabine (as a representative compound that fails to affect P-gp ATPase activity in vitro), alone or in combination (). We found that erlotinib inhibits the exposure of P-gp on the cell surface while having no effects on total P-gp levels (). Immunoblotting-based determinations confirmed the inability of erlotinib to modulate the total levels of P-gp (). Intriguingly, cytarabine was capable of doubling the amount of P-gp exposed at the cell surface (while leaving the total cellular amount of P-gp unaffected), an effect that was entirely blocked in the presence of erlotinib (). Of note, while neither CsA, MK-571 nor verapamil (at various concentrations) significantly reduced the amount of surface-exposed P-gp, erlotinib did so in a dose-dependent fashion ().
Figure 4D–E. In (D and E), quantitative data are reported (P-gp fluorescence normalized to that of DMSO-treated cells or that of the isotype control, means ± SEM, n = 3). **p < 0.01, ***p < 0.001, ns = non-significant (ANOVA plus Bonferroni’s post-hoc test or Dunnett’s test, as appropriate), as compared with DMSO-treated cells stained for surface-exposed P-gp (D and E); ###p < 0.001 (ANOVA plus Bonferroni’s post-hoc test), as compared with cells treated with ARAC only and stained for surface-exposed or P-gp (D). In the inset in (D), immunoblotting data are depicted. β actin levels were assessed to ensure equal loading of lanes. ISO, isotype control; RFI, relative fluorescence intensity.
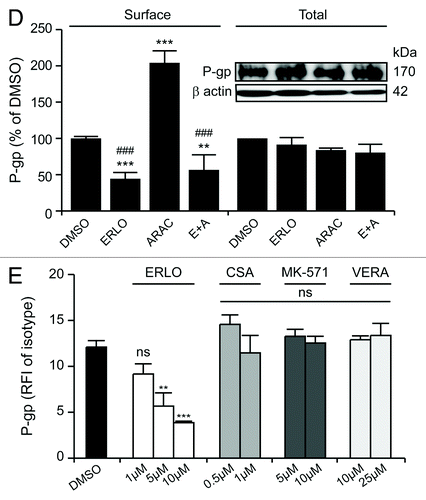
Thus, erlotinib appears to inhibit drub efflux by P-gp by direct altering its enzymatic activity as well as by inhibiting its exposure at the cell surface.
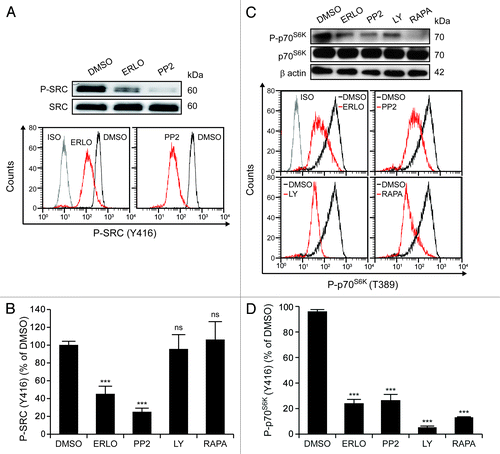
Erlotinib inhibits aberrantly activated SRC kinases and mTOR-conveyed signals
The cell surface exposure and functionality of ABC transporters are modulated by multiple distinct signaling pathways.Citation37 To elucidate whether erlotinib might have an effect on the most prominent of these signaling cascades, we determined the activation status of SRC kinases and mTOR by cytofluorometry and immunoblotting. Short-term (1 h) incubation with erlotinib was sufficient to reduce the constitutive activation of SRC kinases (as monitored by the phosphorylation of tyrosine 416, Y416) in KG-1 cells (). The SRC-specific inhibitor PP2 (but neither the phosphoinositide-3-kinase inhibitor LY294002 nor the mTOR inhibitor rapamycin)Citation38-Citation42 decreased the phosphorylation of Y416 to 20% of baseline levels (). As determined by both immunoblotting and cytofluorometry with a phosphoneoepitope-specific antibody, erlotinib also reduced the phosphorylation of the mTOR substrate p70S6K on threonine 389 (T389), as did PP2, LY294002 and rapamycin ().
Figure 5. Erlotinib inhibits SRC family kinases and mTOR. KG-1 cells were kept in control conditions (DMSO) or incubated with 10 μM erlotinib (ERLO), 10 μM PP2, 10 μM LY294002 (LY) or 10 nM rapamycin (RAPA) for 1 h, then subjected to immunoblotting (A and C) or cytofluorometry (A–D) for the determination of the phosphorylation status of SRC kinases (on Y416) (A and B) or p70S6K (C and D). (A and C) depict immunoblotting results and representative cytofluorometric profiles. β actin levels were monitored to ensure equal lane loading.
To determine whether the inhibition of these signaling pathways translates in reduced MRP-mediated or P-gp-mediated efflux in our model, we evaluated the activity of PP2, LY294002 and rapamycin on the intracellular accumulation of calcein and DiOC2(3), respectively. None of these pathway-specific inhibitors could increase the retention of calcein and DiOC2(3) (Fig. S2A–D), even though all diminished (to a variable extent) the exposure of P-gp on the cell surface (Fig. S2E and F). These data indicate that the pathway-specific inhibition of SRC- and mTOR-conveyed signals fails to recapitulate the effects of erlotinib on the cell surface exposure and functionality of P-gp.
Erlotinib increases chemosensitivity and concomitantly inhibits ABC transporters in patient-derived CD34+ AML cells
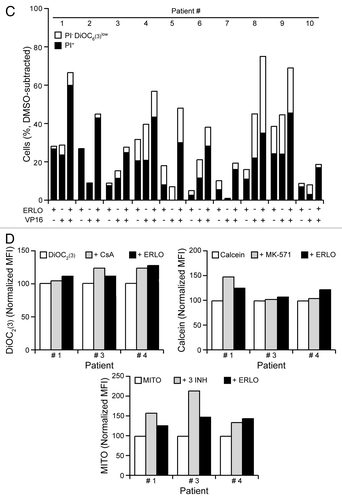
Next, we investigated how erlotinib affects chemosensitivity and drug efflux in primary, patient-derived CD34+ mononuclear cells (). As exemplified in , erlotinib decreased surface-exposed P-gp in CD34+ cells from distinct patients, irrespective of the baseline levels of ecto-P-gp. In line with the effects observed in KG-1 cells (), cytarabine increased the amount of P-gp exposed at the surface of patient-derived CD34+ cells, an effect that was entirely prevented by erlotinib (). Cytofluorometric determinations confirmed that both etoposide and erlotinib can induce the death of malignant CD34+ cells (), although cells from distinct patients exhibited different degrees of sensitivity. When erlotinib and etoposide were combined, we observed variable extents of chemosensitization (), which persisted even in cells that failed to die in response to either compound alone, such as those derived from patient #5 (). Erlotinib exacerbated the intracellular accumulation of DiOC2(3), calcein and mitoxantrone in primary CD34+ AML cells to variable extents, as did the P-gp inhibitor CsA, the MRP inhibitor MK-571 and the combined administration of CsA, KO-143 and MK-571 (). These results indicate that erlotinib is able to hinder drug efflux via multiple ABC transporters in patient-derived cells. Whereas erlotinib increased the percentage of CD34+ cells succumbing from etoposide, neither CsA nor verapamil had similar effects (Fig. S3), underscoring a chemosensitizing potential of erlotinib that may exceed that provided by the inhibition of ABC transporters.
Table 1. Patients’ characteristics
Figure 6A–B. Erlotinib inhibits ABC transporters and enhances chemosensitivity in patient-derived CD34+ cells. (A–C). Patient-derived CD34+ cells were maintained in control conditions (DMSO) or incubated with 5 μM erlotinib (ERLO), 0.1 μM cytarabine (ARAC), or 0.5 μM etoposide (VP16), alone or in combination, for 48 h, then subjected to cytofluorometry for the quantification of or P-glycoprotein (P-gp) exposure on the cell surface (A) or cell death-associated parameters (B and C). (A) depicts representative P-gp exposure results (n = 1). In (B and C), representative dot plots and quantitative data are provided, respectively. The percentage of cells exhibiting mitochondrial transmembrane potential dissipation (PI- DiOC6(3)low) or the breakdown of plasma membrane (PI+) is indicated. Representative results are reported (n = 1).
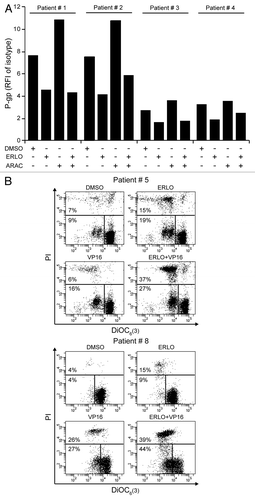
Figure 6C–D. (D). Alternatively, CD34+ cells were loaded with 20 nM DiOC2(3), 10 nM calcein or 0.3 μM mitoxantrone (MITO), alone or combined with 10 μM ERLO, 1 μM cyclosporine A (CsA), 10 μM MK-571 or 1 μM CsA + 0.5 μM KO-143 + 10 μM MK-571 (3 INH) for 2 or 24 h, followed by the cytofluorometric quantification of DiOC2(3) and calcein retention or MITO fluorescence, respectively (D). Data are reported upon normalization to control conditions, as indicated (n = 1). MFI, mean fluorescence intensity.
Discussion
Small molecules inhibiting the EGFR exert antileukemic effects, even though AML cells reportedly do not express the EGFR.Citation13,Citation14,Citation17,Citation19 Thus, EGFR inhibitors employed as standalone agents have been reported to induce complete and durable remission in two leukemia patients that received them owing to a concomitant solid malignancy.Citation16,Citation17 Moreover, we and others have characterized the antileukemic potential of erlotinib and gefitinib in preclinical models, both as standalone interventions and combined with distinct chemotherapeutics.Citation14,Citation15,Citation19,Citation43,Citation44 In particular, we have recently demonstrated that erlotinib and gefitinib synergize with the DNA methyltransferase inhibitor azacytidine in the killing of AML cells in vitro, owing to the fact that both these EGFR inhibitors increase the intracellular accumulation of azacitidine.Citation19
Extending previous observations,Citation20-Citation22 here we show that erlotinib and gefitinib inhibit different types of ABC transporters, including P-gp, BCRP and MRPs, hence facilitating the intracellular accumulation of selected chemotherapeutics and exacerbating their cytotoxicity, either in a synergistic or in an additive fashion. Importantly, at odds with previous studies on this topic, our results were generated in AML cells that express low levels of ABC transportersCitation23-Citation25 and in patient-derived CD34+ cells. Inhibition of ABC pumps by erlotinib and gefinitib was at least partially caused by their ability to inhibit SRC- and mTOR-mediated signaling, which, in turn, limited the exposure of P-gp on the cell surface. In addition, erlotinib was found to mimic verapamil (which is a known inhibitor and substrate of P-gp)Citation36 in its ability to augment the ATPase activity of P-gp. As erlotinib is known to be efficiently extruded by P-gp,Citation45,Citation46 our results indicate that erlotinib operates as a competitive inhibitor of P-gp-mediated efflux.
Increased expression and functionality of ABC transporters are common features of leukemic stem cells and often underlie chemoresistance.Citation3,Citation4,Citation34 Thus, the persistence of AML cells expressing abnormally high levels of ABC transporters is associated with dismal prognosis.Citation5,Citation47 ABC pumps constitute an attractive therapeutic target, as revealed by multiple preclinical and clinical studies showing that inhibitors of drug efflux sensitize AML cells to chemotherapy.Citation5,Citation9 Nevertheless, the clinical use of first-generation P-gp inhibitors (e.g., CsA, verapamil) has been limited by unacceptable side effects.Citation48 Second-generation agents (e.g., PSC833) are better tolerated but interact with other anticancer drugs and are poorly selective for P-gp.Citation48,Citation49 Third-generation inhibitors (e.g., zosuquidar) featuring improved specificity and potency have been developed, but their long-term safety remains to be established.Citation50
One attractive feature of erlotinib—whose short- and long-term toxicity profile is well-studied—is its capacity to simultaneously inhibit different ABC transporters via multiple mechanisms, hence promoting the intracellular accumulation of specific chemotherapeutics. As shown here by us and previously by others,Citation20-Citation22 this greatly exacerbates cytotoxicity, even in cells that do not overexpress ABC pumps and in patient-derived CD34+ cells. Our results do not explain why erlotinib as a standalone agent has been associated with complete and durable regressions in AML patients.Citation16,Citation17 However, based on our data, it is tempting to speculate that the combination of erlotinib and conventional chemotherapeutics may be particularly beneficial for AML patients, irrespective of whether or not their blasts exhibit increased drug efflux via ABC transporters. This hypothesis will have to be formally addressed by carefully designed clinical studies.
Materials and Methods
Patient samples and CD34+ cell selection
Patient samples were studied after obtaining informed consent according to the Declaration of Helsinki. AML and MDS were diagnosed by morphological examination of peripheral blood and bone marrow biopsies, according to the WHO and FAB classifications. Mononuclear cells (MNC) from the peripheral blood or bone marrow were isolated using a Ficoll-Paque PLUS density gradient (Amersham Biosciences). To obtain CD34+ cells from MNCs, a positive selection with the MiniMacs system (Miltenyi Biotec, Bergisch Gladbach) was performed according to the manufacturer’s instructions.
Chemical, cell lines and culture conditions
Unless otherwise indicated, chemicals were purchased from Sigma-Aldrich, while media and supplements for cell culture were purchased from Gibco-Invitrogen. Erlotinib hydrochloride and gefitinib were purchased by LC Laboratories, KO-143 and rapamycin from Tocris Biosciences (Ellisville, USA), verapamil hydrochloride, (4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-α]pyrimidine (PP2) and 2-morpholin-4-yl-8-phenylchromen-4-one (LY294002) from Calbiochem. All chemicals were dissolved in DMSO and stored at -20°C.
Patient-derived CD34+ cells were cultured (37°C, 5% CO2) in Iscove modified Dulbecco medium (IMDM) supplemented with 1% L-glutamine, 100 units/mL penicillin-streptomycin, 10 ng/mL interleukin-3 (Peprotech), 10 ng/mL interleukin-6 (Peprotech), 50 ng/mL thrombopoietin (Peprotech), 100 ng/mL FLT3-ligand (Miltenyi Biotec), 50 ng/mL stem cell factor (SCF) (Miltenyi Biotec) and 20% BIT 9500 serum substitute (200 µg/mL transferrin, 10 µg/mL insulin, 2% bovine serum albumin; from StemCell Technologies). KG-1 and KG-1a cells were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) and cultured in RPMI 1640 medium supplemented with 20% fetal calf serum (FCS). Cells in the logarithmic phase of growth seeded at a density of 1.5 × 105/mL were used for all experiments.
Cytofluorometric assessment of apoptosis
Apoptosis-related parameters were quantified as previously reported.Citation27,Citation51,Citation52 Briefly, live cells were co-stained with 20 nM 3,3′dihexiloxalocarbocyanine iodide [DiOC6(3), from Molecular Probes-Invitrogen], which measures mitochondrial transmembrane potential (Δψm), and 1 μg/mL propidium iodide (PI), which identifies cells with ruptured plasma membrane. Cytofluorometric acquisitions were performed on a FACSCalibur or a FACScan cytofluorometer (BD Biosciences) equipped with a 70 µm nozzle. First-line statistical analysis was performed by means of the CellQuest™ or DIVA 6.1 software (BD Biosciences) upon gating on the events characterized by normal forward scatter and side scatter parameters.
Colorimetric assays
In vitro assessments of the antileukemic effects of etoposide and EGFR inhibitors (alone or in combination) were performed by means of a colorimetric 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)-based assay (CellTiter 96® AQueous one, from Promega) according to the manufacturer’s recommendations. Absorbance at 490 nm was measured by means of a FLUOstar OPTIMA microplate reader (BMG Labtech) and readings were normalized to DMSO-treated cells included in the same test.
Quantification of P-gp expression by cytofluorometry
Cells were harvested, washed once with PBS and pre-incubated with 5 µL/sample FcR blocking reagent (Miltenyi Biotec), to block unwanted binding of antibodies to Fc-receptor expressing cells. Thereafter, cells were co-stained with a phycoerythrin (PE)-conjugated antibody specific for ABCB1 (P-gp) (clone UIC2, Beckman Coulter) and 1 μg/mL PI for 20 min at +4°C (under protection from light), followed by cytofluorometric acquisition. Isotypic PE-conjugated IgG1 from mouse (Beckman Coulter) was used to determine threshold parameters. Dead (PI+) cells were excluded from the analysis.
Immunoblotting
For immunoblotting determinations, cells were lysed on ice in a buffer containing 1% NP40, 20 mM HEPES pH 7.9, 10 mM KCl, 1 mM EDTA, 10% glycerol, 1 mM orthovanadate, 1 mM PMSF, 1 mM dithiothreitol and 10 µg/mL aprotinin, leupeptin, pepstatin followed by centrifugation (20 min, 14,000 rpm) and collection of supernatants. Total protein extracts were then separated on pre-cast 4–12% polyacrylamide gradient gels (Invitrogen) and analyzed following standard immunoblotting proceduresCitation53-Citation55 by using primary antibodies specific for ABCB1/P-gp (Santa Cruz Biotechnology), ABCG2/BCRP (Cell Signaling Technology), β actin (Chemicon), phospho-SRC family (Y416) (Cell Signaling Technology), total SRC (Upstate Biotechnology), phospho-p70S6K (T389) (Cell Signaling Technology) and total p70S6K (Cell Signaling Technology). Primary antibodies were revealed with appropriate horseradish peroxidase-conjugated secondary antibodies (Southern Biotech) and the chemiluminescence detection SuperSignal West Pico® reagent (Thermo Scientific-Pierce).
Efflux studies
Drug efflux via P-gp and MRPs was monitored as previously described,Citation31 with slight modifications. Briefly, cells were pre-incubated with erlotinib (1–15 µM), gefitinib (1–15 µM), cyclosporine A (0.5–1 µM), KO-143 (1 µM), MK-571 (10 µM) or verapamil (10–50 µM) for 15 min to block pump activity and then loaded with fluorescent P-gp and MRP substrates for 1 h. Cells were then washed twice with ice-cold PBS and maintained in the absence or in the presence of the abovementioned pump inhibitors for 1–6 h (to allow for substrate efflux), followed by the cytofluorometric quantification of fluorescence. P-gp efflux studies were performed with 0.5 µg/mL rhodamine 123 (rh123, from Molecular Probes-Invitrogen) or 20 nM 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)]. MRP activity was evaluated with 2.5 µM 5-(and-6)-carboxy-2’,7’diclorofluorescein diacetate (CDCFDA) (Molecular Probes-Invitrogen) or 10 nM calcein AM (Molecular Probes-Invitrogen). Retention values were normalized to the fluorescence of DiOC2(3)- and calcein-labeled cells cultured in the absence of pump inhibitors.
BCRP-mediated efflux (which characterizes the so-called side population) was monitored as previously described,Citation31 with slight modifications. Briefly, KG-1 and KG-1a cells were pre-incubated with 1.5 µM Hoechst 33342 (Molecular Probes-Invitrogen) for 90 min at 37°C with occasional agitation, followed by the administration of 50 µM verapamil, 0.5 µM KO-143 or 10 µM erlotinib for additional 90 min. Thereafter, cells were washed twice with ice-cold PBS, resuspended in PBS supplemented with 1 µg/mL PI and analyzed on a FACSVantage cytofluorometer (BD Biosciences). Hoechst 33342 was excited at 350 nm, and emission was measured at 2 wavelengths (Hoechst blue and Hoechst red) using a 424 bandpass (BP) 44 and a 660 BP 20 optical filter. A 640 nm long-pass filter was used to separate the emission wavelengths.
Evaluation of P-gp ATPase activity
P-gp ATPase activity was evaluated with the luminescence-based Pgp-Glo™ Assay System (Promega) according to the manufacturer’s recommendations. Basal P-gp ATPase activity was determined as the difference between light emission in the presence of sodium orthovanadate (Na3VO4), a selective inhibitor of P-gp, and light emission in control conditions: ΔRLUbasal = RLU(Na3VO4) - RLU(untreated). Similarly, P-gp ATPase activity in the presence of pump inhibitors was determined as follows: ΔRLU(inhibitor) = RLU(Na3VO4) - RLU(inhibitor). For analysis, the ATPase activity in the presence of inhibitors was normalized to the basal activity.
Intracellular phosphoprotein analysis
The phosphorylation of status of p70S6K and SRC was assessed by cytofluorometry, following standard procedures. Briefly, cells were fixed with 1.5% paraformaldehyde for 10 min at room temperature, washed twice with PBS and permeabilized in 1 mL ice-cold 95% methanol for 10 min. Cells were then rehydrated with 5% FCS (v/v in HBSS) for 30 min at 4°C, washed with PBS and incubated for 1 h at 4°C in the dark with primary antibodies specific for phospho-p70S6K (Thr389) or phospho-SRC family (Y416) (both from Cell Signaling Technologies). Rabbit unrelated IgGs (Santa Cruz Biotechnology) were used as isotype controls. Samples stained with the anti-phospho-SRC family antibody were washed twice with PBS and incubated for further 30 min in the dark with secondary goat anti-rabbit AlexaFluor 488 conjugates (Molecular Probes, Invitrogen). Fluorescence was monitored on a Gallios cytofluorometer (Beckman Coulter) and analysis was performed on the Kaluza software (Beckman Coulter). Results were normalized to the mean fluorescence intensity (MFI) of isotype controls and then to the phosphorylation levels displayed by vehicle (DMSO)-treated cells.
Quantification of intracellular etoposide
Etoposide was quantified on a Rapid Resolution Liquid Chromatography (RRLC) 1200SL system coupled to a 6410 Triple Quadripole (QQQ) mass spectrometer (both from Agilent Technologies).Citation56 To this aim, 106 cells were lysed in 1 mL 80% ice-cold methanol (v/v in water), vortexed for 1 min and incubated on ice for 30 min. Lysates were then centrifuged at 13,000 G for 10 min at 4°C, supernatants were dried in a SpeedVac drier (Thermo Scientific-Pierce) and resuspended in 200 μL of 2% methanol (v/v in water) and directly injected into the HPLC-QQQ system. RRLC was performed on 150 × 2.1 mm, 3.5 μM Eclipse Plus columns (Agilent Technologies) with water containing 0.1% formic acid and 7.5 mM ammonium formiate in channel A and methanol in channel B, in gradient mode: t = 0 min, 5% B; t = 10 min, 95% B; t = 12 min; 95% B; re-equilibration time = 6 min. Mass spectrometry was performed in positive electrospray ionization mode at + 4kV on the QQQ system operating in MRM mode. MRM transitions were optimized with direct infusions of etoposide, and two transitions were recorded. The parent ion (MH+ 589.2) at a fragmentor voltage of 180 V gave two daughter ions: one of 229.1 Da at a collision energy of 8 V and one of 185.1 Da at a collision energy of 44 V.
Data treatment and statistical analyses
Unless otherwise specified, experiments were performed in triplicate parallel instances and independently repeated at least twice. Data were analyzed with Excel (Microsoft) and Prism5 (GraphPad Software Inc.). Statistical significance was invariably assessed by means of ANOVA followed by Dunnett’s or Bonferroni’s post-hoc test. p values < 0.05 were considered statistically significant.
| Abbreviations: | ||
| ABC | = | ATP-binding cassette |
| AML | = | acute myeloid leukemia |
| BCRP | = | breast cancer resistance protein |
| CDCFDA | = | 5-(and-6)-carboxy-2’,7’diclorofluorescein diacetate |
| DiOC2(3) | = | 3,3′-diethyloxacarbocyanine iodide |
| DiOC6(3) | = | 3,3′dihexiloxalocarbocyanine iodide |
| EGFR | = | epidermal growth factor receptor |
| MDS | = | myelodysplastic syndrome |
| MFI | = | mean fluorescence intensity |
| MRP | = | multidrug resistance-associated protein |
| P-gp | = | P-glycoprotein |
| PI | = | propidium iodide |
Additional material
Download Zip (3.4 MB)Acknowledgments
We thank David Enot and Maximilien Tailler for precious help with statistical analyses and figure composition, respectively. E.L. receives a fellowship from Post Accueil INSERM, MSe from Fondation de Recherche médicale, S.T. from Institut National du Cancer (INCa). L.G. is funded by the LabEx Immuno-Oncology. G.K. is supported by the Ligue Nationale contre le Cancer (Equipe labelisée), Agence Nationale pour la Recherche (ANR), European Commission (Active p53, Apo-Sys, ChemoRes, ApopTrain), Fondation pour la Recherche Médicale (FRM), INCa, Cancéropôle Ile-de-France, Fondation Bettencourt-Schueller and the LabEx Immuno-Oncology.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Contributions
E.L., M.Se., S.T., M.Sc., C.B. and C.L. performed experiments. E.L. and L.G. wrote the paper and prepared the figures. S.D.B. and P.F. collected clinical samples and provided clinical data. E.L., P.F. and G.K. designed the experimental approach, analyzed the results and wrote the paper.
Related Research Data
References
- Loe DW, Deeley RG, Cole SP. Biology of the multidrug resistance-associated protein, MRP. Eur J Cancer 1996; 32A:945 - 57; http://dx.doi.org/10.1016/0959-8049(96)00046-9; PMID: 8763335
- Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood 2005; 106:1154 - 63; http://dx.doi.org/10.1182/blood-2005-01-0178; PMID: 15870183
- van den Heuvel-Eibrink MM, van der Holt B, Burnett AK, Knauf WU, Fey MF, Verhoef GE, et al. CD34-related coexpression of MDR1 and BCRP indicates a clinically resistant phenotype in patients with acute myeloid leukemia (AML) of older age. Ann Hematol 2007; 86:329 - 37; http://dx.doi.org/10.1007/s00277-007-0269-7; PMID: 17340137
- Wulf GG, Wang RY, Kuehnle I, Weidner D, Marini F, Brenner MK, et al. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood 2001; 98:1166 - 73; http://dx.doi.org/10.1182/blood.V98.4.1166; PMID: 11493466
- Steinbach D, Legrand O. ABC transporters and drug resistance in leukemia: was P-gp nothing but the first head of the Hydra?. Leukemia 2007; 21:1172 - 6; http://dx.doi.org/10.1038/sj.leu.2404692; PMID: 17429427
- Pallis M, Hills R, White P, Grundy M, Russell N, Burnett A. Analysis of the interaction of induction regimens with p-glycoprotein expression in patients with acute myeloid leukaemia: results from the MRC AML15 trial. Blood Cancer J 2011; 1:e23; http://dx.doi.org/10.1038/bcj.2011.23; PMID: 22829167
- Leith CP, Kopecky KJ, Godwin J, McConnell T, Slovak ML, Chen IM, et al. Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood 1997; 89:3323 - 9; PMID: 9129038
- Hirsch P, Tang R, Marzac C, Perrot JY, Fava F, Bernard C, et al. Prognostic impact of high ABC transporter activity in 111 adult acute myeloid leukemia patients with normal cytogenetics when compared to FLT3, NPM1, CEBPA and BAALC. Haematologica 2012; 97:241 - 5; http://dx.doi.org/10.3324/haematol.2010.034447; PMID: 22058196
- List AF, Kopecky KJ, Willman CL, Head DR, Persons DL, Slovak ML, et al. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood 2001; 98:3212 - 20; http://dx.doi.org/10.1182/blood.V98.12.3212; PMID: 11719356
- Shtil AA, Azare J. Redundancy of biological regulation as the basis of emergence of multidrug resistance. Int Rev Cytol 2005; 246:1 - 29; http://dx.doi.org/10.1016/S0074-7696(05)46001-5; PMID: 16164965
- Settleman J. Inhibition of mutant EGF receptors by gefitinib: targeting an Achilles’ heel of lung cancer. Cell Cycle 2004; 3:1496 - 7; http://dx.doi.org/10.4161/cc.3.12.1325; PMID: 15539955
- Ji H, Sharpless NE, Wong KK. EGFR targeted therapy: view from biological standpoint. Cell Cycle 2006; 5:2072 - 6; http://dx.doi.org/10.4161/cc.5.18.3277; PMID: 16969107
- Stegmaier K, Corsello SM, Ross KN, Wong JS, Deangelo DJ, Golub TR. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood 2005; 106:2841 - 8; http://dx.doi.org/10.1182/blood-2005-02-0488; PMID: 15998836
- Boehrer S, Adès L, Braun T, Galluzzi L, Grosjean J, Fabre C, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood 2008; 111:2170 - 80; http://dx.doi.org/10.1182/blood-2007-07-100362; PMID: 17925489
- Boehrer S, Adès L, Galluzzi L, Tajeddine N, Tailler M, Gardin C, et al. Erlotinib and gefitinib for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a preclinical comparison. Biochem Pharmacol 2008; 76:1417 - 25; http://dx.doi.org/10.1016/j.bcp.2008.05.024; PMID: 18617157
- Chan G, Pilichowska M. Complete remission in a patient with acute myelogenous leukemia treated with erlotinib for non small-cell lung cancer. Blood 2007; 110:1079 - 80; http://dx.doi.org/10.1182/blood-2007-01-069856; PMID: 17644748
- Pitini V, Arrigo C, Altavilla G. Erlotinib in a patient with acute myelogenous leukemia and concomitant non-small-cell lung cancer. J Clin Oncol 2008; 26:3645 - 6; http://dx.doi.org/10.1200/JCO.2008.17.0357; PMID: 18640945
- Komrokji RS, Lancet JE, Yu D, Santana E, Yan L, Smith PS, et al. Erlotinib for treatment of myelodysplastic syndromes: A phase II clinical study. Blood 2010; 116
- Lainey E, Wolfromm A, Marie N, Enot D, Scoazec M, Bouteloup C, et al. Azacytidine and erlotinib exert synergistic effects against acute myeloid leukemia. Oncogene 2012; In press
- Kitazaki T, Oka M, Nakamura Y, Tsurutani J, Doi S, Yasunaga M, et al. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 2005; 49:337 - 43; http://dx.doi.org/10.1016/j.lungcan.2005.03.035; PMID: 15955594
- Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res 2007; 67:11012 - 20; http://dx.doi.org/10.1158/0008-5472.CAN-07-2686; PMID: 18006847
- Noguchi K, Kawahara H, Kaji A, Katayama K, Mitsuhashi J, Sugimoto Y. Substrate-dependent bidirectional modulation of P-glycoprotein-mediated drug resistance by erlotinib. Cancer Sci 2009; 100:1701 - 7; http://dx.doi.org/10.1111/j.1349-7006.2009.01213.x; PMID: 19493273
- Bailly JD, Muller C, Jaffrézou JP, Demur C, Gassar G, Bordier C, et al. Lack of correlation between expression and function of P-glycoprotein in acute myeloid leukemia cell lines. Leukemia 1995; 9:799 - 807; PMID: 7769842
- Steinbach D, Sell W, Voigt A, Hermann J, Zintl F, Sauerbrey A. BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia. Leukemia 2002; 16:1443 - 7; http://dx.doi.org/10.1038/sj.leu.2402541; PMID: 12145683
- Amico D, Barbui AM, Erba E, Rambaldi A, Introna M, Golay J. Differential response of human acute myeloid leukemia cells to gemtuzumab ozogamicin in vitro: role of Chk1 and Chk2 phosphorylation and caspase 3. Blood 2003; 101:4589 - 97; http://dx.doi.org/10.1182/blood-2002-07-2311; PMID: 12576328
- Farag SS, Ruppert AS, Mrózek K, Mayer RJ, Stone RM, Carroll AJ, et al. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: a cancer and leukemia group B study. J Clin Oncol 2005; 23:482 - 93; http://dx.doi.org/10.1200/JCO.2005.06.090; PMID: 15534356
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16:1093 - 107; http://dx.doi.org/10.1038/cdd.2009.44; PMID: 19373242
- Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis 2007; 12:803 - 13; http://dx.doi.org/10.1007/s10495-007-0720-1; PMID: 17294081
- Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10:221 - 37; http://dx.doi.org/10.1038/nrd3373; PMID: 21358741
- Harbron C. A flexible unified approach to the analysis of pre-clinical combination studies. Stat Med 2010; 29:1746 - 56; http://dx.doi.org/10.1002/sim.3916; PMID: 20572122
- Walter RB, Pirga JL, Cronk MR, Mayer S, Appelbaum FR, Banker DE. PK11195, a peripheral benzodiazepine receptor (pBR) ligand, broadly blocks drug efflux to chemosensitize leukemia and myeloma cells by a pBR-independent, direct transporter-modulating mechanism. Blood 2005; 106:3584 - 93; http://dx.doi.org/10.1182/blood-2005-02-0711; PMID: 16051742
- Feller N, Broxterman HJ, Währer DC, Pinedo HM. ATP-dependent efflux of calcein by the multidrug resistance protein (MRP): no inhibition by intracellular glutathione depletion. FEBS Lett 1995; 368:385 - 8; http://dx.doi.org/10.1016/0014-5793(95)00677-2; PMID: 7628644
- Tsimberidou AM, Paterakis G, Androutsos G, Anagnostopoulos N, Galanopoulos A, Kalmantis T, et al. Evaluation of the clinical relevance of the expression and function of P-glycoprotein, multidrug resistance protein and lung resistance protein in patients with primary acute myelogenous leukemia. Leuk Res 2002; 26:143 - 54; http://dx.doi.org/10.1016/S0145-2126(01)00106-0; PMID: 11755464
- Guo Y, Lübbert M, Engelhardt M. CD34- hematopoietic stem cells: current concepts and controversies. Stem Cells 2003; 21:15 - 20; http://dx.doi.org/10.1634/stemcells.21-1-15; PMID: 12529547
- Koeffler HP, Billing R, Lusis AJ, Sparkes R, Golde DW. An undifferentiated variant derived from the human acute myelogenous leukemia cell line (KG-1). Blood 1980; 56:265 - 73; PMID: 6967340
- Lan ZJ, Tang YM, Shen HQ, Qian BQ, Ning BT, Chen YH. [Evaluation of the P-gp pump function on leukemic cell membrane and proper application of its reversal agents with Calcein-AM and flow cytometry]. Zhonghua Er Ke Za Zhi 2007; 45:334 - 8; PMID: 17697617
- Hirose M. Biology and modulation of multidrug resistance (MDR) in hematological malignancies. Int J Hematol 2002; 76:Suppl 2 206 - 11; http://dx.doi.org/10.1007/BF03165119; PMID: 12430927
- Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011; 10:4230 - 6; http://dx.doi.org/10.4161/cc.10.24.18486; PMID: 22107964
- Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle 2010; 9:1859 - 62; http://dx.doi.org/10.4161/cc.9.10.11872; PMID: 20436272
- Cozzi M, Giorgi F, Marcelli E, Pentimalli F, Forte IM, Schenone S, et al. Antitumor activity of new pyrazolo[3,4-d]pyrimidine SRC kinase inhibitors in Burkitt lymphoma cell lines and its enhancement by WEE1 inhibition. Cell Cycle 2012; 11:1029 - 39; http://dx.doi.org/10.4161/cc.11.5.19519; PMID: 22333592
- Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle 2009; 8:1896 - 900; http://dx.doi.org/10.4161/cc.8.12.8809; PMID: 19478560
- Xu R, Spencer VA, Groesser DL, Bissell MJ. Laminin regulates PI3K basal localization and activation to sustain STAT5 activation. Cell Cycle 2010; 9:4315 - 22; http://dx.doi.org/10.4161/cc.9.21.13578; PMID: 20980837
- Lindhagen E, Eriksson A, Wickström M, Danielsson K, Grundmark B, Henriksson R, et al. Significant cytotoxic activity in vitro of the EGFR tyrosine kinase inhibitor gefitinib in acute myeloblastic leukaemia. Eur J Haematol 2008; 81:344 - 53; PMID: 18637032
- Boehrer S, Galluzzi L, Lainey E, Bouteloup C, Tailler M, Harper F, et al. Erlotinib antagonizes constitutive activation of SRC family kinases and mTOR in acute myeloid leukemia. Cell Cycle 2011; 10:3168 - 75; http://dx.doi.org/10.4161/cc.10.18.16599; PMID: 21897118
- de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs 2012; 30:443 - 9; http://dx.doi.org/10.1007/s10637-010-9569-1; PMID: 20963470
- Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MA, Beijnen JH, et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther 2008; 7:2280 - 7; http://dx.doi.org/10.1158/1535-7163.MCT-07-2250; PMID: 18723475
- Schaich M, Soucek S, Thiede C, Ehninger G, Illmer T, SHG AML96 Study Group. MDR1 and MRP1 gene expression are independent predictors for treatment outcome in adult acute myeloid leukaemia. Br J Haematol 2005; 128:324 - 32; http://dx.doi.org/10.1111/j.1365-2141.2004.05319.x; PMID: 15667534
- Coley HM. Overcoming multidrug resistance in cancer: clinical studies of p-glycoprotein inhibitors. Methods Mol Biol 2010; 596:341 - 58; http://dx.doi.org/10.1007/978-1-60761-416-6_15; PMID: 19949931
- van der Holt B, Löwenberg B, Burnett AK, Knauf WU, Shepherd J, Piccaluga PP, et al. The value of the MDR1 reversal agent PSC-833 in addition to daunorubicin and cytarabine in the treatment of elderly patients with previously untreated acute myeloid leukemia (AML), in relation to MDR1 status at diagnosis. Blood 2005; 106:2646 - 54; http://dx.doi.org/10.1182/blood-2005-04-1395; PMID: 15994288
- Tang R, Faussat AM, Perrot JY, Marjanovic Z, Cohen S, Storme T, et al. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer 2008; 8:51; http://dx.doi.org/10.1186/1471-2407-8-51; PMID: 18271955
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 2010; 1:e10; http://dx.doi.org/10.1038/cddis.2009.8; PMID: 21364612
- de La Motte Rouge T, Galluzzi L, Olaussen KA, Zermati Y, Tasdemir E, Robert T, et al. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res 2007; 67:6253 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-07-0538; PMID: 17616683
- Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res 2010; 70:1793 - 803; http://dx.doi.org/10.1158/0008-5472.CAN-09-3112; PMID: 20145152
- Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16:1006 - 17; http://dx.doi.org/10.1038/cdd.2009.34; PMID: 19325567
- Criollo A, Galluzzi L, Maiuri MC, Tasdemir E, Lavandero S, Kroemer G. Mitochondrial control of cell death induced by hyperosmotic stress. Apoptosis 2007; 12:3 - 18; http://dx.doi.org/10.1007/s10495-006-0328-x; PMID: 17080328
- Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, et al. Prognostic impact of vitamin b6 metabolism in lung cancer. Cell Rep 2012; 2:257 - 69; http://dx.doi.org/10.1016/j.celrep.2012.06.017; PMID: 22854025