Abstract
Here we report that miR-93, a miRNA in the miR-106B~25 cluster, a paralog of the miR-17–92 cluster, was significantly upregulated in human breast carcinoma tissues. We stably expressed miR-93 in the MT-1 human breast carcinoma cell line and found that tumors formed by the miR-93 cells contained more blood vessels than those formed by the control cells. Co-culture experiments indicated that the MT-1 cells displayed a high activity of adhesion with endothelial cells and could form larger and more tube-like structures with endothelial cells. Lung metastasis assays were performed in a mouse metastatic model, and it was found that expression of miR-93 promoted tumor cell metastasis to lung tissue. In cell culture, expression of miR-93 enhanced cell survival and invasion. We examined the potential target that mediated miR-93’s effects and found that the large tumor suppressor, homology 2 (LATS2) was a target of miR-93. Higher levels of LATS2 were associated with cell death in the tumor mass. Silencing LATS2 expression promoted cell survival, tube formation and invasion, while ectopic expression of LATS2 decreased cell survival and invasion. These findings demonstrated that miR-93 promoted tumor angiogenesis and metastasis by suppressing LATS2 expression. Our results suggest that the inhibition of miR-93 function may be a feasible approach to repress tumor metastasis.
Keywords: :
Introduction
MicroRNAs (miRNA) are single-stranded, non-coding RNAs, 18 to 25 nucleotides in length. They are transcribed from genomic DNA to make long primary transcripts, which are modified by RNase III-type enzymes Drosha and Dicer to produce precursor miRNAs and then mature miRNAs.Citation1 More than 1,000 miRNAs have been detected in human cells. Mature miRNAs can bind to the complementary sequences in the 3′-untranslated regions (3′UTR) of target mRNAs,Citation2 resulting in post-transcriptional repression. On the other hand, the 3′UTR has been shown to regulate miRNA functions.Citation3-Citation5 As a new class of regulatory molecules, miRNAs have diverse functions in regulating cell activities associated with cell proliferation,Citation6-Citation8 differentiation,Citation9 invasion,Citation10 tissue morphogenesis and growth,Citation11,Citation12 tumor formation,Citation13-Citation15 angiogenesisCitation16-Citation19 and metastasis.Citation20-Citation22 The largest functional group of miRNAs are the ones involved in cancer development, and among these, some have been reported to function as oncogenic miRNAs or tumor suppressors, while others exert diverse functions.Citation23-Citation25 A primary transcript usually consists of a miRNA cluster that gives rise to multiple precursors and mature miRNA.Citation26 These miRNAs can form polycistronic clusters or exist individually.
One of the most intensively studied clusters is miR-17~92, which has paralogs, miR-106A~363 and miR-106B~25, that play important roles in cancer development through the repression of many tumor-associated genes.Citation27-Citation30 The overexpression of miR-17~92 enhances cell proliferation and reduces apoptosis by regulating cell cycle progression.Citation31-Citation33 The oncogenic functions of mir-17~92, mir-106A~363 and mir-106B~25 have been extensively reported.Citation31,Citation34-Citation36 Most recently, mir-106B~25 cluster was reported to exert oncogenic effects in hepatocellular carcinoma.Citation37 However, the precise functions of each miRNA in the mir-106B~25 cluster are not clear. This cluster of miRNAs contains three pre-miRNAs: mir-106B, mir-93 and mir-25. Interestingly, miR-106B and miR-93 share identical seed regions, suggesting that these two miRNAs may exert the prevailing functions in this cluster. Previous studies indicated that miR-93 can repress the tumor suppressor TP53INP1 in human T-cell leukemia virus 1-transformed human T-cellsCitation38 and FUS-1 in human lung cancer cell lines.Citation39 We have also found that expression of miR-93 promoted tumor growth and angiogenesis in a human brain tumor cell model.Citation16 To study the role of miR-93 in other types of cancers, we analyzed levels of miR-93 in human breast carcinoma specimens and found significant upregulation of miR-93 in the tumor tissues. This study was designed to explore the function of miR-93 in breast cancer angiogenesis and metastasis.
Results
MiR-93 affects the interaction of tumor and endothelial cells and angiogenesis
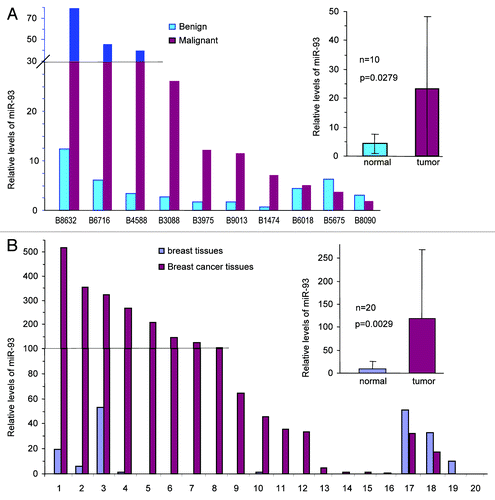
We have previously reported that the human brain tumor cell line, U87, transfected with miR-93 can grow faster and form more blood vessels in nude mice.Citation16 In this study, we studied the roles of miR-93 in breast cancer development. RNAs were isolated from paraffin-embedded tumor tissues and benign tissues of patients with breast carcinoma. Analysis of miR-93 with real-time PCR indicated that there was a significant upregulation of miR-93 levels in the tumor tissues compared with benign tissues (, p = 0.0279). We also isolated RNAs from the tumors and adjacent benign tissues of 20 patients that were lymph node-positive, an indication of metastasis. Analysis of miR-93 in these patients showed significant higher levels of miR-93 in the tumor tissues than the benign tissues (, p = 0.0029).
Figure 1. Expression of miR-93 in human breast carcinoma specimens. (A) RNAs were isolated from paraffin blocks of human breast carcinoma specimens and the benign breast tissues, followed by real-time PCR analysis of miR-93 levels. The breast carcinoma tissues expressed significantly higher levels of miR-93 than the benign tissues. (B) RNAs were isolated from paraffin blocks of human breast carcinoma specimens with lymph-positive (metastasis) and the benign breast tissues followed by analysis of miR-93 levels. The tumor tissues expressed significantly higher levels of miR-93.
To study how miR-93 might affect breast cancer development, we stably transfected breast carcinoma cell line MT-1 with a miR-93 expression construct or a control vector that harbored a random sequence. The cells were injected into CD-1 nude mice. Tumors excised from the mice were cut into sections. To evaluate angiogenesis or the blood vessel density, the tumor sections were immunohistochemically stained for CD34 expression, which served as a marker for the blood vessel endothelium. It was noted that tumors formed by the miR-93-expressing cells showed a greater number of blood vessels than the mock tumors (, upper). The blood vessels were counted in four randomly selected fields for each cell type and a significant difference was obtained (, lower).
Figure 2. Expression of miR-93 enhanced breast cancer angiogenesis. (A) Top: sections of tumor from mice injected with miR-93- or mock-MT-1 cells were probed with CD34 primary rat antibody in 10% goat serum with TBS, followed by probing with anti-rat IgG (positive). The negative or control tumor sections were probed with anti-rat polyclonal IgG antibody only. There were increased levels of tumor-associated vascularization (as shown with arrows), where the miR-93 expression in MT-1 cells was enhanced. Scale bars, 50 µm. Bottom: the average count of the number of the blood vessels found in the tumor sections at four randomly selected fields. Error bars, SD (n = 4), **p < 0.0001. (B) Mock- and miR-93-transfected cells were co-cultured with endothelial cells YPEN or EOMA or lung cells BEAS-2B at the ratio of 2:1. After 2 d of culture, the co-cultured cells were photographed. The miR-93 cells displayed higher capacities in expansion than the vector-transfected cells. As a result, the endothelial cells were squeezed into small islands by the miR-93 cells. The miR-93 cells could mix well with the lung cells compared with the mock control. (C) MT-1 cells stably transfected with miR-93 or mock were seeded on tissue culture plates at a cell density of 1.5 x 105 cells/well on 6-well plates. After overnight culture, endothelial cells YPEN were inoculated on top of the existing cultures (6 x 104 cells/well). After an additional overnight culture, the endothelial cells were able to spread over the miR-93 cultures, but could not spread over the mock cultures. (D) The miR-93- and mock-transfected cells were mixed with YPEN cells and inoculated in Matrigel, followed by examination of tube formation. The YPEN cells formed larger complexes and longer tubes when mixed with the miR-93 expressing cells compared with the mock-transfected cells.
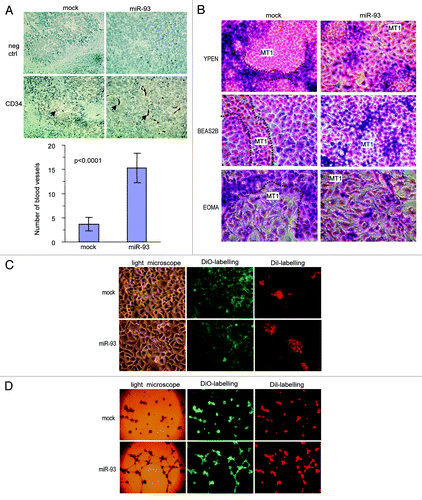
To understand how expression of miR-93 affected angiogenesis, we tested the effect of miR-93 on endothelial cell activities. The miR-93- or mock-transfected cells were mixed and co-cultured with rat endothelial YPEN cells. Two to three days after cell inoculation, the cultures were fixed and stained for examination of cell morphology. It was found that the mock-transfected MT-1 cells did not interact with YPEN cells: each group of cells tended to group together. On the other hand, the miR-93 MT-1 cells could mix and interact well with the YPEN cells (). The YPEN cells linked together or squeezed into stretches when co-cultured with the mock MT-1 cells, while the cells were squeezed into small island-like nodules when co-cultured with the miR-93 MT-1 cells (Fig. S1A). When the mock and miR-93 MT-1 cells were co-cultured with mouse endothelial cells EOMA, similar results were obtained (; Fig. S1B). We also co-cultured the mock and miR-93 MT-1 cells with human bronchial epithelial cells BEAS-2B, and found that the miR-93 MT-1 cells could mix well with the BEAS-2B cells (; Fig. S1C), suggesting stronger cell-cell interactions.
MT-1 cells stably transfected with miR-93 or mock vectors were seeded on tissue culture plates at different cell densities overnight followed by inoculation of endothelial cells YPEN on top of the existing cultures. The mixed cultures were examined under a light and fluorescent microscope. We observed that at lower cell densities of MT-1 cells, endothelial cells tended to attach to empty areas in the mock culture plates. However, endothelial cells could adhere well to the miR-93-transfected MT-1 cells. At higher MT-1 cell densities, endothelial cells attached on top of the MT-1 cell cultures. After an additional overnight culture, the endothelial cells were able to spread on the miR-93 cultures, but not on the mock-transfected cells (; Fig. S2A)
The miR-93- or mock-transfected cells were also mixed with the YPEN cells and cultured in Matrigel to examine tube formation. In the presence of the miR-93-transfected cells, YPEN cells formed larger complexes and longer, tube-like structures compared with the mock-transfected cells (; Fig. S2B). These results indicated that miR-93-expressing MT-1 cells supported endothelial cell activities.
MiR-93 enhances breast cancer metastasis
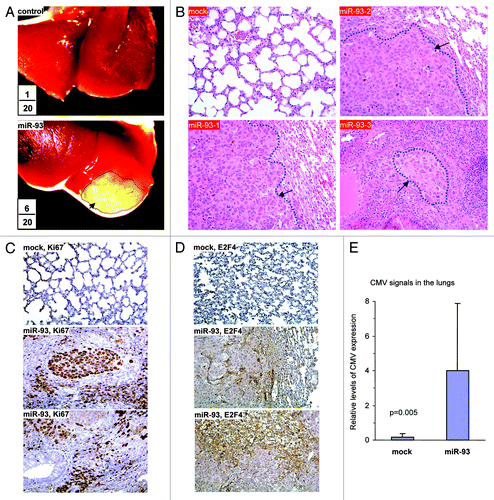
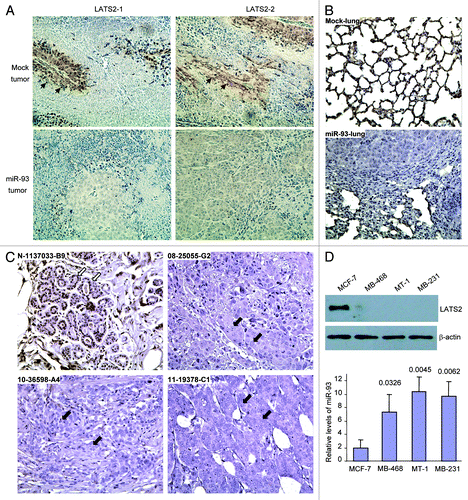
We then tested the role of miR-93 in a different mouse model by injecting the cells into NOD-SCID mice via the tail vein. Seven weeks after the injection, the mice were sacrificed and examined. It was noted that five mice in the miR-93 group developed cachectic lung tumors. Typical metastatic lesions in the lungs are shown (). The numbers in the figure were combined from two injections, indicating the number of mice injected and the number of mice developing lung tumors. Lung sections were obtained and subjected to H&E staining. Nodules were detected, indicating metastasis of the tumor cells to the lungs (). Lung sections were also analyzed for Ki67 reactivity, a marker of proliferation. It was found that the miR-93 tumor displayed greater number of Ki67-positive cells than the control (). The sections were also probed for E2F4, a tumor suppressor. Since the antibody used was specific for human E2F4, we could only detect positive cells in the tumor sections but not in the mouse lung sections. Interestingly, the E2F4-positive cells were either unhealthy or were localized in the areas showing extensive cell death (). DNA was isolated from lung tissues and subjected to PCR to amplify the CMV promoter, which was used to drive the expression of miR-93 in the construct, to indicate metastasis of the cancer cells. Expression of miR-93 significantly increased CMV levels (). The level of CMV signal in each mouse was also provided (Fig. S2C).
Figure 3. MiR-93 enhanced breast cancer metastasis to the lung. (A) Mock- or miR-93-transfected MT-1 cells (2 × 105) were injected into the tail vein of CD-1 nude mice (n = 20). Six weeks after the injection, six mice in the miR-93 group developed visible tumors in the lungs, but only one in the control group. Typical metastatic lesions in the lungs are shown (arrows). (B) H&E staining of lungs from mock and miR-93 mice showed metastasis lesions in the miR-93 lungs (arrows). (C) The sections were immunohistochemically stained with antibody against Ki67. The miR-93 tumor sections showed higher levels of Ki67 staining than the control group. (D) The sections were also probed with antibody against E2F4. The miR-93 tumor sections showed E2F4 staining, which was not detected in the control group. (E) DNA was isolated from lung tissues and subjected to PCR to amplify the CMV promoter to indicate metastasis of the tissues. Expression of miR-93 promoted metastasis.
MiR-93 promotes tumor cell survival and invasion
The in vivo data suggest that miR-93 may enhance tumor cell survival and invasion. We therefore performed in vitro studies to test this possibility. MT-1 cells transfected with miR-93 or mock were seeded in tissue culture dishes or Petri dishes. The cells were maintained in serum-free conditions. Transfection with miR-93 enhanced cell survival compared with the controls both in tissue culture dishes and Petri dishes (; Fig. S3A). We then conducted cell invasion assays. Mock- and miR-93- transfected MT-1 cells were loaded into the insert with serum-free medium and then incubated at 37°C for 24 h. The invasive cells were stained and counted in six randomly selected fields under a light microscope. We found that cells transfected with miR-93 displayed higher levels of invasion than the mock-transfected cells (; Fig. S3B).
Figure 4. MiR-93 promotes cell survival and invasion. (A) The MT-1 cells transfected with miR-93 or mock (2.5 x 105 cells per 1 mL) were seeded onto Petri dishes and tissue culture dishes and were maintained in a serum-free medium at 37°C and 5% CO2 for 7 d. The cells were counted at the end of seventh day. The miR-93 transfected cells were seen to survive better. Error bars indicated standard deviation (SD) with n = 5. (B) The miR-93 and mock cells (1 x 105) suspended in 100 μl serum-free medium were loaded onto the insert and incubated at 37°C for 48 h. The invasive cells were stained blue and were counted in six fields of views/membrane using a light microscope. Error bars indicate SD (n = 6). (C) MT-1 cells were transiently transfected with anti-miR-93 oligos or control oligos with random sequence. The cultures were maintained in tissue culture dishes in serum-free DMEM for 5 d, followed by microscopic examination and photographed. The number of cells was counted for statistical analysis. Error bars indicate SD (n = 6). (D) MT-1 cells were transiently transfected with anti-miR-93 oligos or control oligos with random sequence or with anti-miR-93 plasmid or control vector. The cells (1 x 105) were subjected to invasion assays at 37°C for 64 h. Transfection with anti-miR-93 inhibited cell invasion.
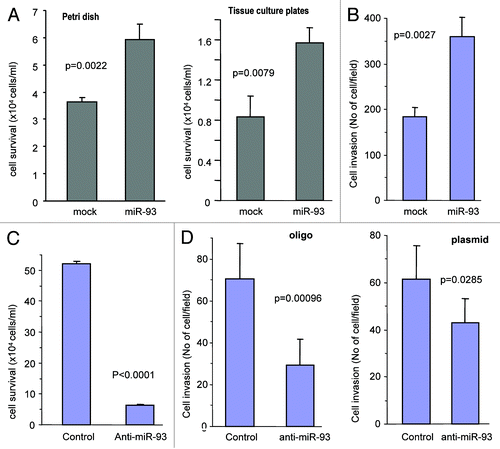
To confirm the function of miR-93, we transfected MT-1 cells with a construct expressing an antisense sequence against miR-93 (anti-miR-93) followed by culturing for 5 d. Cell survival assays showed a significant reduction in survival rate in the miR-93-expressing cells transfected with anti-miR-93 as compared with the control (; Fig. S3C). We also performed invasion assays, and found that expression of anti-miR-93 significantly decreased the activity of invasion (; Fig. S3D).
MiR-93 represses LATS2 expression
We investigated the target of miR-93 in mediating the observed effects focusing on tumor suppressors. The large tumor suppressor homology 2 (LATS2 or KPM) was identified as a potential target of miR-93. The 3′UTR of LATS2 harbored two typical target sequences for miR-93 at nucleotides 3955–3977 and nucleotides 4058–4078 (). Cell lysates prepared from the miR-93- and mock-transfected cells were analyzed on protein gel blot probed with anti-LATS2 antibody. We found that expression of LATS2 was repressed in the miR-93 transfected cells ().
Figure 5. Targeting analysis of LATS2 by miR-93. (A) Computational analysis indicated that miR-93 potentially targeted LATS2 located at nucleotides 3955–3977 and nucleotides 4058–4078. (B) Cell lysate prepared from miR-93- or mock-transfected MT-1 cells was analyzed on protein gel blot for LATS2 expression. LATS2 expression was repressed by miR-93 transfection. Staining for β-actin from the same membrane confirmed equal loading. (C) Top: two luciferase constructs were generated, each containing a fragment harboring the target site of miR-93, producing Luc-Lats-3955 and Luc-Lats-4057. Mutations were also generated on the potential target sequence (red color), resulting in two mutant constructs Luc-Lats-3955-mut and Luc-Lats-4057-mut. Bottom: MT-1 cells were co-transfected with miR-93 and a luciferase reporter construct. The luciferase reporter vector was used as a control. Asterisks indicate significant differences. Error bars, SD (n = 3). (D) Top: alignment of the miR-93 target sites on LATS2 located at nucleotides 3955–3977 across Homo sapiens (NM_014572), Pan troglodytes (XM_509566), Bos taurus (XM_584953), Mus musculus (NM_015771), Gallus gallus (XM_417143), Xenopus (Silurana) tropicalis (NM_001102704), Rattus norvegicus (NM_001107267) and Taeniopygia guttata (XM_002191071). The seed regions for miR-93-LATS2 interactions are boxed. Bottom: conservation of the sequences is shown across all species. (E) Top: alignment of the miR-93 targeting LATS2 located at nucleotides 4058–4078 across the same species. The seed regions for miR-93-LATS2 interactions are boxed. Bottom: conservation of the sequences is shown across all species.
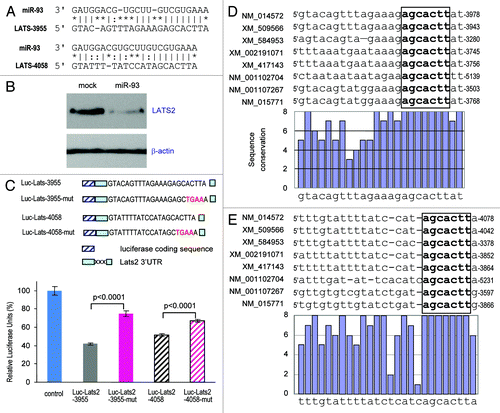
To obtain direct evidence that the 3′UTR of LATS2 was a target of miR-93, we generated two luciferase expression constructs harboring fragments of the LATS2 3′UTR containing the miR-93 target sites, which produced the constructs Luc-Lats-3955 and Luc-Lats-4058 (, upper; Fig. S4A). Two mutant constructs Luc-Lats-3955-mut and Luc-Ltas-4058-mut were also generated (, upper; Fig. S4B). Expression of Luc-Lats-3955 and Luc-Lats-4058 showed that luciferase activities were significantly repressed when the constructs were co-transfected with miR-93 (, lower). Mutations of the miR-93 target sites abolished the effects of miR-93. Examination of the target sequences indicated that the miR-93 target sites at nucleotides 3955–3977 () and nucleotides 4058–4078 () were highly conserved across different species. In all species obtained, the seed regions that were critical for miR-93 targeting were 100% homologous.
Sections from the miR-93 and mock tumors were probed with anti-LATS2 antibody. In the miR-93 tumor sections, LATS2 levels were much lower than the mock tumor sections (). Interestingly, LATS2 expression was co-localized with cell death in both mock and miR-93 tumor sections. The downregulation of LATS2 expression was also seen in the miR-93-metastatic lung sections compared with the stromal tissues (). We also examined LATS2 levels in human breast carcinoma specimens. Tumor sections from a number of randomly picked patients were immunostained with anti-LATS2 antibody. We could only detect LATS2 expression in the benign breast tissues, but not in the tumor mass (; Fig. S5A).
Figure 6. Repression of LATS2 expression in the miR-93 tumors. (A) Sections of tumor from mice injected subcutaneously with miR-93- or mock-MT-1 cells were probed with LATS2 primary goat antibody in 10% goat serum, followed by probing with anti-goat IgG. There were higher levels of LATS2 staining (arrows) in the mock tumor section compared with the miR-93 tumor sections. (B) To examine the metastatic tumors, sections from the lungs were probed with anti-LATS2 antibody. The levels of LATS2 were lower in the miR-93 group than in the control group. (C) Sections from human breast carcinoma and normal tissues (N) were subjected to immunohistochemistry for LATS2 expression. LATS2 was detected in the duct structure (open arrows) but not in the tumor mass (closed arrows). (D) Top: Cell lysates prepared from different human breast cancer cell lines were subjected to protein gel blot analysis probed with anti-LATS2 and anti-actin antibodies. LATS2 was detected in the benign breast cell line MCF-7. Right: the levels of miR-93 were analyzed by real-time PCR. The MCF-7 cells expressed significantly lower levels of miR-93 than the other breast cancer cell lines.
To test whether or not LATS2 expression was correlated with miR-93 levels, a number of human breast cancer cell lines were analyzed for LATS2 expression by protein gel blot analysis, and miR-93 levels were analyzed by real-time PCR (Fig. S6). We found that only the benign breast cell line MCF-7 expressed detectable LATS2 (). Consistent with this, the MCF-7 cells expressed the lowest level of miR-93. Similar results were obtained by using mouse breast cancer cell lines (Fig. S5B).
Confirmation of the targeting effects by silencing and rescue experiments
To confirm that LATS2 played an important role in mediating miR-93’s effects in regulating cell survival and invasion, we introduced siRNA to suppress expression of LATS2. MT-1 cells were transfected with four siRNAs targeting LATS2 and maintained in serum-free medium. Cells transfected with the siRNAs displayed increased survival rates as compared with cells transfected with the control oligo (; Fig. S6A).
Figure 7. LATS2 mediates the effects of miR-93. (A) The MT-1 cells (1 × 105 cells/ml/well) were transfected with siRNA oligos targeting LATS2 or a control oligo. The cells were seeded onto a 12-well plate and incubated in serum-free medium at 37°C for 7 days. The survived cells were counted. Transfection with siRNAs increased cell survival. Data are mean ± SD (n = 4). (B) The MT-1 cells transfected with siRNA oligos targeting LATS2 or a control oligo were subjected to invasion assays at 37°C for 48 h. Transfection with anti-miR-93 inhibited cell invasion. Transfection with siRNAs increased invasion. Error bars, SD (n = 4). (C) Ectopic expression of LATS2 in the miR-93 cells reversed miR-93 effect on cell survival. (D) The miR-93-expressing cells were transfected with LATS2 and a control vector. The cells were suspended in 100 ml serum-free medium and loaded in the transwell insert containing Matrigel, followed by incubation at 37°C for 60 h for invasion assays. Transfection with LATS2 partially reversed the effect of miR-93 on cell invasion. (E) Ectopic expression of LATS2 in the miR-93 cells reversed miR-93 effect on tube formation.
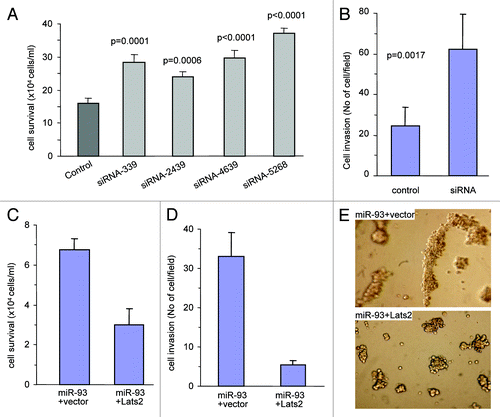
Since all siRNAs appeared to have functioned efficiently, one of them, the siRNA-2439, was used for further analysis. MT-1 cells transfected with siRNA-2439 were subjected to invasion assays. It was found that the siRNA-2439-transfected cells showed a higher ability in invading the Matrigel than the control oligo-transfected cells (; Fig. S6B). Cell lysates prepared from MT-1 cells transfected with siRNA-2439 or a control oligo were analyzed on protein gel blot to confirm the silencing effect of the siRNA targeting LATS2 (Fig. S6C). These results suggested that the LATS2-mediated pathway was essential for miR-93-enhanced cell survival and invasion.
To confirm that miR-93 promoted cell survival and invasion by targeting LATS2, rescue experiments were performed. MT-1 cells stably transfected with miR-93 were transfected with LATS2 expression construct or a control vector. Confirmation of the expression of LATS2 was assayed on protein gel blot probed with anti-LATS2 antibody (Fig. S6D). Survival assay showed that reintroduction of LATS2 into the miR-93-expressing cells reversed the effect of miR-93 on cell survival (; Fig. S6E). In cell invasion assays, decreased invasion was found in cells transfected with LATS2 (; Fig. S6F). In tube formation assays, we found that ectopic expression of LATS2 decreased tube formation (), suggesting its role in reducing angiogenesis. Thus, re-expression of LATS2 was sufficient to cause cell death, and decrease both cell invasion and tube formation. This suggested that the effects of miR-93 on enhanced angiogenesis and metastasis were at least partly taking place through repression of LATS2 expression.
Discussion
MiR-93 is expressed in the miR-106B~25 cluster, along with other miRNAs such as miR-106B and miR-25. Recent studies have demonstrated that the miR-106B~25 cluster is involved in oncogenic activities. Consistent with other groups, we also reported that the expression of miR-93 promotes tumor growth.Citation16,Citation39 The seed region of miR-93 (nucleotides 2–8 of the miRNA) is identical to miR-106B in the same cluster, and to miR-17, miR-20a, miR-20b, and miR-106A in two paralogs (miR-17~92 and miR-106A~363 clusters) of the miR-106B-25 cluster. Since the seed regions are the critical sites for gene targeting, it suggests that these six miRNAs could target the same mRNAs and play similar roles. Indeed, these three paralogs have been reported to play a critical role in cancer development.Citation16,Citation31,Citation40
In this study, we utilized two model systems to study the roles of miR-93 in breast cancer development: a local tumor formation assay to examine angiogenesis and a metastasis assay to analyze tumor invasion and metastasis to the lungs. In the local tumor formation assay, although expression of miR-93 did not enhance tumor growth, it promoted angiogenesis significantly. The miR-93 tumor sections showed much more blood vessel formation than the mock tumor sections. This could be clearly seen in the sections with H&E staining and the slides stained for CD34, a marker of blood vessels. These results suggested that miR-93 tumor cells promoted angiogenesis, similar to the results we reported previously.Citation16
The function of miR-93 in the promotion of angiogenesis was corroborated with in vitro experiments. We found that miR-93 enhanced endothelial cell activities, including cell spreading and tube formation. In the cell-cell interaction experiments, YPEN endothelial cells were able to spread over the miR-93-transfected cells better than on the mock-transfected cells. These results suggested that the surface of the miR-93-transfected cells were different from that of the mock-transfected cells. MiR-93 may have repressed cell surface proteins. We have previously demonstrated that miR-93 could repress integrin β8,Citation16 allowing the spreading of endothelial cells and close contact of endothelial cells to miR-93-transfected cells. These would favor the process of blood vessel extension or angiogenesis.
To understand how expression of miR-93 affected angiogenesis, we performed tube-formation and co-culture experiments, and found that miR-93-transfected cells displayed greater activity in forming tube-like structures than the mock-transfected cells when co-cultured with YPEN cells. The formation of longer and more complex tubes in Matrigel by YPEN cells co-cultured with the miR-93-transfected MT-1 cells was an indication of enhanced angiogenesis. When the cell number was low, no extensive tubes were formed. Larger complexes were seen in the presence of miR-93-expressing cells. This result further confirmed that the miR-93-transfected cells were able to interact well with endothelial cells facilitating blood vessel formation.
In co-culture experiments, the miR-93- and mock-transfected MT-1 cells were mixed with YPEN or EOMA endothelial cells. In both cases, the mock MT-1 cells did not interact well with either of the endothelial cells. However, the miR-93 MT-1 cells could adhere well with both types of endothelial cells. This indicated that the miR-93 MT-1 cells allowed extension of the endothelial cells into the tumor mass. In other words, the miR-93 MT-1 cells could facilitate angiogenesis of the endothelial cells. These results may partly explain why the miR-93 tumors contained much more vasculature than the mock tumors.
In tumor metastasis experiments, we found that the miR-93-transfected cells had a greater activity in metastasis to the lung than the mock-transfected cells. As the nuclei of the MT-1 breast cancer cells appeared much larger than the lung cells, the cancer cells could be readily distinguished from the stromal cells. Morphologically, the large complex of the tumor mass also allowed identification of the metastatic mass compared with the surrounding stroma. In addition, the stromal cells displayed a limited capacity in cell proliferation, as shown by weak Ki67 staining. On the contrary, the metastatic MT-1 cells displayed strong Ki67 immunoreactivity, an indication of extensive cell proliferation. Interestingly, we detected significant levels of Ki67-positive cells with small nuclei, mixing with the tumor cells that had large nuclei. This may have been the consequence of inflammatory cell infiltration of the tumors. We also observed that some areas in the tumor mass had no Ki67-positive cells. This may have been a consequence of cell death, since necrosis typically accompanies tumor growth, due to lack of nutrition. Corroboration of these results were obtained by immunohistochemical analysis of E2F4, a tumor cell death-associated molecule. We detected extensive staining of E2F4 in the metastatic tumor mass.
To understand how expression of miR-93 promoted tumor metastasis, we performed tumor cell survival and invasion assays. By cell survival assays, we found that the miR-93-transfected MT-1 cells were able to survive significantly longer compared with the mock-transfected MT-1 cells when maintained in serum-free conditions. Since the miR-93-expressing cells did not grow significantly faster than the controls, the promotion of cell survival did not appeared as a consequence of tumor cell growth. To allow metastasis to occur, single cancer cells or a limited number of cancer cells must be able to survive while invading to a new location. The ability of miR-93 cells to survive may play an important role in the metastasis of cancer cells.
MiR-93 appeared to play roles in enhancing invasion of MT-1 cells. The miR-93-MT-1 cells were able to penetrate through the Matrigel as compared with the control cells. The invasiveness of transformed cells represents an essential step in tumor progression. The transition from tumor growth to metastatic disease is defined by the ability of tumor cells from the primary site to invade local tissues and cross tissue barriers. To initiate the metastatic process, cancer cells must first penetrate the epithelial basement membrane. They invade the interstitial stroma by active proteolysis of the dense matrix containing collagens, glycoproteins and proteoglycans, among others. Degradation of these matrix molecules allows cancer cells to break through the established adhesion barriers of the local tissues. It appeared that the miR-93 MT-1 cells were able to digest the matrix molecules, which facilitated cell invasion and led to metastasis.
In order to understand how miR-93 functioned, it was essential to identify the target of miR-93 in our experimental system. Since expression of miR-93 promoted angiogenesis and metastasis, the target(s) were thought to be a tumor suppressor or may have functioned as negative regulators of angiogenesis and metastasis. Computational analysis showed that many potential targets of miR-93, were associated with tumor growth, including the large tumor suppressor homolog 2 (LATS2/KPM).
LATS2 is a member of the LATS tumor suppressor family. It plays a central role in the Hippo pathway in the inhibition of cell growth and in tumor suppression.Citation41,Citation42 Other proteins in the Hippo pathway can trigger LATS2 activation, resulting in the inhibition of cell growth.Citation43,Citation44 LATS2 can regulate mitotic progression and p53 activity, leading to suppression of tumor growth.Citation45,Citation46 In addition, LATS2 plays roles in regulating Retinoblastoma protein (pRB) activity associated with senescence, cell cycle arrest and inhibition of tumor growth.Citation47 Our experiments demonstrated that expression of miR-93 repressed LATS2 levels, leading to the promotion of angiogenesis and metastasis. Since the effects of LATS2 on tumor angiogenesis and metastasis are not known, we confirmed that LATS2 played important roles in mediating miR-93 functions associated with angiogenesis and metastasis by a series of experiments. Silencing LATS2 expression by siRNA indicated that LATS2 played important roles in cell survival and invasion. In rescue experiments, we demonstrated that ectopic expression of LATS2 decreased cell survival, cell invasion and tumor formation, all of which were associated with angiogenesis and metastasis. Nevertheless, further studies are needed to dissect the signal pathways underlying the effects of LATS2 on angiogenesis and metastasis.
In summary, we have demonstrated that miR-93 functions as an oncogene by enhancing tumor cell survival, blood vessel formation and tumor metastasis by targeting LATS2. MiR-93 can potentially target a great number of genes, some acting directly on tumorigenesis and angiogenesis. Others may only indirectly affect tumorigenesis and angiogenesis. Thus, we cannot exclude the possibility that other miR-93 targeting proteins may also play a role in the system we studied. Future investigation would involve elucidating the network by which miR-93 functions and understanding its contribution to cancer development.
Materials and Methods
Materials and cell culture
The monoclonal antibodies against LATS2 were purchased from Santa Cruz Biotechnology. The monoclonal antibody against β-actin used was obtained from Sigma. Horseradish peroxidase-conjugated goat anti-mouse IgG and horseradish peroxidase-conjugated goat anti-rabbit IgG were obtained from Bio-Rad. Immunoblotting was performed using the ECL protein gel blot detection kit. High Pure PCR Template Preparation Kits were obtained from Roche Applied Science. The breast cancer MT-1 cell line was used in this experiment. The cells were cultured in RPMI-1640 supplemented with 10% fetal-bovine serum, FBS (Invitrogen). These cells were maintained in an incubator at 37°C under an atmosphere of 5% CO2. The culture medium was changed twice a week. The LATS2 expression construct is a kind gift from Dr. Nicholas Dyson in Massachusetts General Hospital Cancer Center, Harvard Medical School.Citation47
Construct generation
A cDNA sequence containing two human precursor miR-93 units was inserted into a mammalian expression vector pEGFP-N1 in the restriction enzyme sites BglII and HindIII. The sequence of the precursor miR-93 in the construct is the same as the endogenous sequence.
We used a luciferase reporter vector (pMir-Report, Ambion) to generate luciferase reporter constructs. There are two potential binding sites for miR-93 in the LATS2 3′UTR. These two sites are close to each other, which made the analysis of each site individually difficult. It is essential to develop a strategy to distinguish the effect of each site separately. The LATS2 fragment was cloned with two primers HuLats2-R93-SacI (5′ gg gagctc tagatgggggccaggcacccccac) and HuLats2-R93-MluI (5′mL acgcgt cct aaa tgtg aat aagt gct atgg). The PCR product was digested with SacI and MluI, followed by insertion into a SacI- and MluI-opened pMir-Report vector. To generate a mutant containing a mutation in the second site of the 3′UTR (leaving the other site active), the primer HuLats2-R93-SacI was combined with a primer HuLATS2-R93-MluI-mut2 (5′mL acgcgt cct aaa tgtg aat ttca gct) in a PCR, followed by cloning of the fragment in the reporter vector. To generate a mutant containing mutations in both sites, the primer HuLats2-R93-SacI was combined with the primer HuLATS2-R93-MluI-mut (5′mL acgcgt cct aaa tgtg aat ttca gct atgg ataaaatacaaa tgtagaaaataacagcagca tgattt gtcaaa gtt aat ccc tat aattta gtaa gaaaaaa tgg atat aaac aaaa t ttca gctc) in a PCR. To generate a mutant containing a mutation in the first site, the primers HuLats2-R93-SacI and HuLats2-R93-MluI were used in a PCR with the construct containing both mutation sites as a template. Thus, all inserts had exactly the same size for comparison in luciferase activity assays.
Clinical specimens
Breast carcinoma specimens were collected at the time of mastectomy from previously untreated patients. Primary tumors and surrounding normal breast tissues were freshly excised and fixed in 10% formalin overnight, immersed in 70% ethanol, embedded in paraffin and sectioned. The sections were subjected to immunohistochemistry probed with antibodies against LATS2. The work was conducted following a protocol approved by the Clinical Research Ethics Committee at the University of Iowa Carver College of Medicine. The paraffin-fixed specimens were also used to isolated miRNAs. In brief, small RNAs were isolated from formalin-fixed, paraffin-embedded tissue blocks, using the Total Nucleic Acid Isolation Kit (Ambion) or from freshly frozen tissues. Quantification of miRNAs was performed with real-time PCR.
Cell invasion assay
Cell invasion assay was performed with the modified chemotactic Boyden chamber assays as described.Citation48 Briefly, cells were loaded into 8-μm cell culture inserts and placed in 24-well cell culture plates. The upper chamber of the Polyethylene Terephthalate (PET) membrane was coated with 100 μl diluted Matrigel (1 mg/ml). The lower chamber was filled with 600 μl 10% FBS/DMEM medium. Cell suspension (100 µL containing 3 × 105 cells) was transferred to the upper chamber. The transwell was incubated at 37°C for 4 h. Cells (1 × 105) in 100 μl serum-free DMEM medium were gently loaded onto each filter insert (upper chamber) and then incubated at 37°C for 24–72 h. The filter inserts were removed from the chambers, fixed with methanol for 5 min and stained with Harris’ hemotoxylin for 20 min. Samples were subsequently washed, dried and mounted onto slides. The invasive cells were stained blue and visualized under a microscope (Axiover Inverted Microscope, Zeiss) then counted in six random fields for statistical analysis.
Cell survival assay
The method was described in detail previously.Citation49 In brief, cells (1.5 × 105 cells per well or 2 × 105 cells per well) were seeded on 35-mm Petri dishes or 12-well tissue culture dishes in DMEM containing 0–10% FBS and maintained at 37°C for 12 h. After cell attachment, the medium was changed to serum-free DMEM medium or 10% FBS/DMEM medium. Cells were harvested daily, and cell number was determined by a Coulter Counter.
Co-culture experiments
MiR-93- or mock-transfected MT-1 cells were mixed with stromal cells, YPEN, BEAS-2B or EOMA (1.5 × 105 cells/ml for each) and seeded on 3.5-cm culture dishes in DMEM supplemented with 10% FBS (2 ml). The morphology of two types of cells was examined under a light microscope daily. The images were captured at different intervals.
In YPEN cell-spreading experiments, the miR-93- or mock-transfected MT-1 cells were cultured at different cell densities (0.5–1.5 × 105 cells/well) in tissue culture plates overnight. Next day, YPEN cell were inoculated on top of the MT-1 cell cultures (6 × 104 cells/well). YPEN cell spreading on top of the MT-1 cells was examined under a light and fluorescent microscope.
To test the effect of miR-93 on tube formation, we mixed the miR-93- or mock-transfected MT-1 cells with YPEN cells. The mixture was cultured in Matrigel. The interaction of both types of cells and the formation of tube-like structures were examined under a light and fluorescent microscope.
Transfection of MT-1 cells with siRNAs
The siRNAs were designed and synthesized by GenePharma. Four siRNAs that were synthesized to target LATS2 expression were used in this experiment: siRNA-1 (LATS2–2439) sense 5′cuauguuugucaagaucaatt; siRNA-2 (LATS2–4369) sense 5′gcagauuucuucuauuauatt; siRNA-3 (LATS2–339) sense 5′gaaaguauguuuacaacaatt; siRNA-4 (LATS2–5268) sense 5′gaaguuuaucaguguuuaatt; and control sense 5′uucuccgaacgcgucacgutt. siRNAs transfection was performed using Lipofectamine 2000 (Invitrogen). In brief, 4 × 104 cells in 2 mL of RPMI-1640 (10%FBS) were plated in each of five different 35-mm tissue culture dishes and were incubated overnight at 37°C and 5% CO2 atmosphere. For each dish, 10 µL siRNA was added into 150 µL of serum-free medium and mixed with 3 µL of Lipofectamine. The mixture was added to cells and incubated for 6 h before replacing the medium with RPMI-1640 containing 10% FBS. Total protein assay was prepared 72 h after transfection for protein gel blot analysis.
Real-time PCR
Total RNAs were extracted from cell cultures with mirVana miRNA Isolation Kit according to the instructions of the manufacturer (Ambion). Real-time PCR was performed as previously described.Citation50 For mature miRNA analysis, the total RNAs were extracted from ~1 × 106 cells. The first strand cDNA was synthesized using 1 μg RNA. Real-time PCR was performed with QuantiMir-RT Kit using 1 μl cDNA as template. To perform these experiments, other kits were also needed, including Qiagen, miScript Reverse Transcription Kit, cat#218060, miScript Primer Assay, cat#218411, and miScriptSYBR GreenPCR Kit, cat#218073. The primer specific for mature miR-93 was purchased from Qiagen. The primers used as real-time PCR controls were human-U6RNAf and human-U6RNAr as described.Citation12
Protein gel blot analysis
To prepare cell lysates, the miR93- or mock-transfected MT-1 cells were seeded onto five different 35-mm cultures dish at a density of 1.5 × 105 cells/plate. When the culture reached sub-confluence, the cells in each dish were lysed with 100 µL of lysis buffer containing protease inhibitors (150 mM NaCl, 25 mM TRIS-HCl, pH 8.0, 0.5 M EDTA, 20% Triton X-100, 8 M urea, and 1x protease inhibitor cocktail). Protein concentration was measured by Bio-Rad Protein Assay Kit (#5000–0006). The lysates were subjected to SDS-PAGE (sodium dodecyl sulfate PAGE). After being separated, the proteins were transferred onto a nitrocellulose membrane followed by immunostaining with anti-LATS2 primary antibody at the dilution of 1:400 at 4°C overnight. The following day, this membrane was washed in TBST (Tris-Buffered saline and Tween 20) and then incubated with HRP-conjugated goat-anti-mouse secondary antibody for 2 h at room temperature. The protein bands were detected by enhanced chemiluminescence (ECL) detection. The blot was re-probed with anti-β-actin mouse primary antibody and goat-anti-mouse secondary antibody to confirm equal loading of samples.
Luciferase activity assay
Luciferase activity assay was performed by using a dual-luciferase reporter system developed by Promega (E1960). The cells were cultured on 12-well tissue culture plates at a density of 2 × 105 cells per well in DMEM containing 10% FBS. The cultures were maintained at 37°C for 24 h, until sub-confluence. The cultures were co-transfected with the luciferase reporter constructs, corresponding miRNA mimics and Renilla luciferase construct (as an internal control) mixed with Lipofectamine 2000. The cells were then collected and lysed with 150 µl of passive lysis buffer per well from Luciferase Assay Kit (Promega) on a shaker for 20 min. The cell lysates was centrifuged at 3,000 rpm for 5 min. The supernatants were transferred into a black 96-well plate (3 × 10 μl) for luciferase activity measurement and into a transparent 96-well plate (3 × 50 μl) for Renilla activity analysis. For the luciferase activity measurement, luciferase assay reagent (70 μl) was added to each well, and the luciferase activities were detected by using microplate scintillation and luminescence counter (Packard, Perkin Elmer). For the internal control of Renilla activities, 90 μl of assay reagent (4 mg/ml ONPG, 0.5M MgSO4, β-mercaptoethanol and 0.4 M sodium phosphate buffer) were added to each well. The plate was then incubated at 37°C for 60 min. Renilla luciferase activity was then measured in the same tube. The absorbance at 410 nm was measured by using a microplate reader (Bio-Tek Instruments, Inc.). Luciferase activities between different treatments were compared after normalization with Renilla luciferase activities.
Tumorigenicity assays in nude mice
Tumor formation assay was performed as previously described.Citation51 In brief, 5-week-old CD1 strain nude mice were injected with the miR-93- or mock-transfected MT-1 cells at the cell number of 5 × 106 cells in 150 μl PBS per mouse. Tumor sizes were measured weekly thereafter. When the sizes of the tumors were above the limit described by the animal protocol approved by the Animal Care Committee at Sunnybrook Research Institute, the mice were sacrificed and the tumors were removed. Tumors were fixed in 10% buffered formalin (Histochoice Tissue Fixative MB, Amresco), processed and embedded in paraffin. Immunohistochemistry was performed on 5-μm paraffin sections mounted on charged slides. The sections were stained with H&E, and immunostained with CD34 to detect blood vessels. Sections were also immunostained for expression of LATS2.
Lung metastasis in nude mice
The mock- and miR-93-tansfected MT-1 cells were cultured in 10% FBS/RPMI 1640 media at 37°C with 5% CO2 until sub-confluence. The cells were given fresh 10% FBS/RPMI 1640 media 24 h before being harvested for injection into the mice. Cell viability was determined, and cells were suspended with greater than 95% viability without cell clumping. Five-week-old CD-1 nude mice were injected via tail vein using the above transfected cells (2 × 105 cells in 150 μl 10% FBS/ RPMI 1640 medium). Mice were grouped at random, 10 mice in each group. All mice were sacrificed seven weeks after injection. At necroscopy, lungs, liver, spines were dissected and examined carefully. Half the organs were fixed in 10% formalin, and the other half were frozen in liquid nitrogen for subsequent analysis.
To analyze metastasis, mouse lung tissues were homogenized, and the genomic DNAs were isolated with High Pure PCR Template Preparation Kit according to the instructions of the manufacturer. Tumor burden for each individual tissue was measured using real-time PCR. Primers used were as follows: CMV forward (5′gtcatcgctattaccatggtgatgcgg) and CMV reverse (5′agctctgcttatatagacctcccaccg) for genotyping; β-actin forward (5′ccggcatgtgcaaagccggcttcg) and β-actin reverse (5′ctcattgtagaaggtgtggtgcc) for loading control.
Tissue processing, H&E staining and immunohistochemistry
Primary tumors or lungs were freshly excised and fixed in 10% formalin. The fixed organs were subsequently immersed in 70% ethanol, embedded in paraffin and sectioned. The sections were subjected to H&E staining and immunohistochemistry. Briefly, tissue sections were de-paraffinized with xylene and ethanol and then boiled in a pressure cooker. After washing with Tris-Buffered-Saline (TBS) containing 0.025% Triton X-100, the sections were blocked with 10% goat serum for 1 h. The sections were then incubated with primary antibody in TBS containing 10% goat serum albumin overnight. The sections were washed and labeled with biotinylated secondary antibody, followed by avidin conjugated horseradish peroxidase provided by the Vectastain ABC kit (Vector, PK-4000). The slides were subsequently stained with DAB followed by Mayer's Hematoxylin for counter staining and slide mounting.
Statistical analysis
The results (mean values ± SD) of all the experiments were subjected to statistical analysis by t-test. The level of significance was set at p < 0.05, p < 0.01 and p < 0.0001.
| Abbreviations: | ||
| DMEM | = | Dulbecco’s modified Eagle’s medium |
| FBS | = | fetal bovine serum |
| PCR | = | polymerase chain reaction |
| PAGE | = | polyacrylamide gel electrophoresis |
| GFP | = | green fluorescent protein |
| siRNA | = | small interfering RNA |
| LATS2 (or KPM) | = | the large tumor suppressor homology 2 |
Additional material
Download Zip (13.9 MB)Acknowledgments
This work was supported by grants from Canadian Institutes of Health Research (MOP-102635 and MOP-111171) to Burton B. Yang who is the recipient of a Career Investigator Award (CI 7418) from the Heart and Stroke Foundation of Ontario. W.Y. was a recipient of NSERC Undergraduate Summer Research Award.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003; 425:415 - 9; http://dx.doi.org/10.1038/nature01957; PMID: 14508493
- Lai EC. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 2002; 30:363 - 4; http://dx.doi.org/10.1038/ng865; PMID: 11896390
- Lee DY, Shatseva T, Jeyapalan Z, Du WW, Deng Z, Yang BB. A 3′-untranslated region (3’UTR) induces organ adhesion by regulating miR-199a* functions. PLoS One 2009; 4:e4527; http://dx.doi.org/10.1371/journal.pone.0004527; PMID: 19223980
- Jeyapalan Z, Deng Z, Shatseva T, Fang L, He C, Yang BB. Expression of CD44 3′-untranslated region regulates endogenous microRNA functions in tumorigenesis and angiogenesis. Nucleic Acids Res 2011; 39:3026 - 41; http://dx.doi.org/10.1093/nar/gkq1003; PMID: 21149267
- Lee SC, Fang L, Wang CH, Kahai S, Deng Z, Yang BB. A non-coding transcript of nephronectin promotes osteoblast differentiation by modulating microRNA functions. FEBS Lett 2011; 585:2610 - 6; http://dx.doi.org/10.1016/j.febslet.2011.07.016; PMID: 21784074
- Viticchiè G, Lena AM, Latina A, Formosa A, Gregersen LH, Lund AH, et al. MiR-203 controls proliferation, migration and invasive potential of prostate cancer cell lines. Cell Cycle 2011; 10:1121 - 31; http://dx.doi.org/10.4161/cc.10.7.15180; PMID: 21368580
- Shatseva T, Lee DY, Deng Z, Yang BB. MicroRNA miR-199a-3p regulates cell proliferation and survival by targeting caveolin-2. J Cell Sci 2011; 124:2826 - 36; http://dx.doi.org/10.1242/jcs.077529; PMID: 21807947
- Hidaka H, Seki N, Yoshino H, Yamasaki T, Yamada Y, Nohata N, et al. Tumor suppressive microRNA-1285 regulates novel molecular targets: aberrant expression and functional significance in renal cell carcinoma. Oncotarget 2012; 3:44 - 57; PMID: 22294552
- Kahai S, Lee SC, Lee DY, Yang J, Li M, Wang CH, et al. MicroRNA miR-378 regulates nephronectin expression modulating osteoblast differentiation by targeting GalNT-7. PLoS One 2009; 4:e7535; http://dx.doi.org/10.1371/journal.pone.0007535; PMID: 19844573
- Luo L, Ye G, Nadeem L, Fu G, Yang BB, Honarparvar E, et al. MicroRNA-378a-5p promotes trophoblast cell survival, migration and invasion by targeting Nodal. J Cell Sci 2012; 125:3124 - 32; http://dx.doi.org/10.1242/jcs.096412; PMID: 22454525
- Wang CH, Lee DY, Deng Z, Jeyapalan Z, Lee SC, Kahai S, et al. MicroRNA miR-328 regulates zonation morphogenesis by targeting CD44 expression. PLoS One 2008; 3:e2420; http://dx.doi.org/10.1371/journal.pone.0002420; PMID: 18560585
- Shan SW, Lee DY, Deng Z, Shatseva T, Jeyapalan Z, Du WW, et al. MicroRNA MiR-17 retards tissue growth and represses fibronectin expression. Nat Cell Biol 2009; 11:1031 - 8; http://dx.doi.org/10.1038/ncb1917; PMID: 19633662
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 2006; 103:2257 - 61; http://dx.doi.org/10.1073/pnas.0510565103; PMID: 16461460
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 2006; 20:2202 - 7; http://dx.doi.org/10.1101/gad.1444406; PMID: 16882971
- Nohata N, Hanazawa T, Enokida H, Seki N. microRNA-1/133a and microRNA-206/133b clusters: dysregulation and functional roles in human cancers. Oncotarget 2012; 3:9 - 21; PMID: 22308266
- Fang L, Deng Z, Shatseva T, Yang J, Peng C, Du WW, et al. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-β8. Oncogene 2011; 30:806 - 21; http://dx.doi.org/10.1038/onc.2010.465; PMID: 20956944
- Zou C, Xu Q, Mao F, Li D, Bian C, Liu LZ, et al. MiR-145 inhibits tumor angiogenesis and growth by N-RAS and VEGF. Cell Cycle 2012; 11:2137 - 45; http://dx.doi.org/10.4161/cc.20598; PMID: 22592534
- Lee DY, Deng Z, Wang CH, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A 2007; 104:20350 - 5; http://dx.doi.org/10.1073/pnas.0706901104; PMID: 18077375
- Smits M, Nilsson J, Mir SE, van der Stoop PM, Hulleman E, Niers JM, et al. miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget 2010; 1:710 - 20; PMID: 21321380
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007; 449:682 - 8; http://dx.doi.org/10.1038/nature06174; PMID: 17898713
- Huang Q, Gumireddy K, Schrier M, le Sage C, Nagel R, Nair S, et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol 2008; 10:202 - 10; http://dx.doi.org/10.1038/ncb1681; PMID: 18193036
- Rutnam ZJ, Yang BB. The involvement of microRNAs in malignant transformation. Histol Histopathol 2012; 27:1263 - 70; PMID: 22936445
- Ory B, Ellisen LW. A microRNA-dependent circuit controlling p63/p73 homeostasis: p53 family cross-talk meets therapeutic opportunity. Oncotarget 2011; 2:259 - 64; PMID: 21436470
- Yang X, Rutnam ZJ, Jiao C, Wei D, Xie Y, Du J, et al. An anti-let-7 sponge decoys and decays endogenous let-7 functions. Cell Cycle 2012; 11:3097 - 108; http://dx.doi.org/10.4161/cc.21503; PMID: 22871741
- Yang W, Lee DY, Ben-David Y. The roles of microRNAs in tumorigenesis and angiogenesis. Int J Physiol Pathophysiol Pharmacol 2011; 3:140 - 55; PMID: 21760972
- Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 2005; 33:2697 - 706; http://dx.doi.org/10.1093/nar/gki567; PMID: 15891114
- Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell 2008; 132:860 - 74; http://dx.doi.org/10.1016/j.cell.2008.02.020; PMID: 18329371
- Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem 2007; 282:2135 - 43; http://dx.doi.org/10.1074/jbc.M608939200; PMID: 17135249
- Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008; 133:217 - 22; http://dx.doi.org/10.1016/j.cell.2008.04.001; PMID: 18423194
- Bonauer A, Dimmeler S. The microRNA-17-92 cluster: still a miRacle?. Cell Cycle 2009; 8:3866 - 73; http://dx.doi.org/10.4161/cc.8.23.9994; PMID: 19887902
- Matsubara H, Takeuchi T, Nishikawa E, Yanagisawa K, Hayashita Y, Ebi H, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene 2007; 26:6099 - 105; http://dx.doi.org/10.1038/sj.onc.1210425; PMID: 17384677
- Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65:9628 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-05-2352; PMID: 16266980
- Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle 2005; 4:1179 - 84; http://dx.doi.org/10.4161/cc.4.9.2032; PMID: 16096373
- Landais S, Landry S, Legault P, Rassart E. Oncogenic potential of the miR-106-363 cluster and its implication in human T-cell leukemia. Cancer Res 2007; 67:5699 - 707; http://dx.doi.org/10.1158/0008-5472.CAN-06-4478; PMID: 17575136
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008; 132:875 - 86; http://dx.doi.org/10.1016/j.cell.2008.02.019; PMID: 18329372
- Dang CV. MYC, microRNAs and glutamine addiction in cancers. Cell Cycle 2009; 8:3243 - 5; http://dx.doi.org/10.4161/cc.8.20.9522; PMID: 19806017
- Li Y, Tan W, Neo TW, Aung MO, Wasser S, Lim SG, et al. Role of the miR-106b-25 microRNA cluster in hepatocellular carcinoma. Cancer Sci 2009; 100:1234 - 42; http://dx.doi.org/10.1111/j.1349-7006.2009.01164.x; PMID: 19486339
- Yeung ML, Yasunaga J, Bennasser Y, Dusetti N, Harris D, Ahmad N, et al. Roles for microRNAs, miR-93 and miR-130b, and tumor protein 53-induced nuclear protein 1 tumor suppressor in cell growth dysregulation by human T-cell lymphotrophic virus 1. Cancer Res 2008; 68:8976 - 85; http://dx.doi.org/10.1158/0008-5472.CAN-08-0769; PMID: 18974142
- Du L, Schageman JJ, Subauste MC, Saber B, Hammond SM, Prudkin L, et al. miR-93, miR-98, and miR-197 regulate expression of tumor suppressor gene FUS1. Mol Cancer Res 2009; 7:1234 - 43; http://dx.doi.org/10.1158/1541-7786.MCR-08-0507; PMID: 19671678
- Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 2009; 23:2806 - 11; http://dx.doi.org/10.1101/gad.1872909; PMID: 20008931
- Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle 2010; 9:3892 - 903; http://dx.doi.org/10.4161/cc.9.19.13386; PMID: 20935475
- Li Y, Pei J, Xia H, Ke H, Wang H, Tao W. Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene 2003; 22:4398 - 405; http://dx.doi.org/10.1038/sj.onc.1206603; PMID: 12853976
- Paramasivam M, Sarkeshik A, Yates JR 3rd, Fernandes MJ, McCollum D. Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol Biol Cell 2011; 22:3725 - 33; http://dx.doi.org/10.1091/mbc.E11-04-0300; PMID: 21832154
- Cornils H, Kohler RS, Hergovich A, Hemmings BA. Downstream of human NDR kinases: impacting on c-myc and p21 protein stability to control cell cycle progression. Cell Cycle 2011; 10:1897 - 904; http://dx.doi.org/10.4161/cc.10.12.15826; PMID: 21593588
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev 2006; 20:2687 - 700; http://dx.doi.org/10.1101/gad.1447006; PMID: 17015431
- Yabuta N, Mukai S, Okada N, Aylon Y, Nojima H. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle 2011; 10:2724 - 36; http://dx.doi.org/10.4161/cc.10.16.16873; PMID: 21822051
- Tschöp K, Conery AR, Litovchick L, Decaprio JA, Settleman J, Harlow E, et al. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev 2011; 25:814 - 30; http://dx.doi.org/10.1101/gad.2000211; PMID: 21498571
- Rutnam ZJ, Yang BB. The non-coding 3′ UTR of CD44 induces metastasis by regulating extracellular matrix functions. J Cell Sci 2012; 125:2075 - 85; PMID: 22637644
- Wu QP, Xie YZ, Deng Z, Li XM, Yang W, Jiao CW, et al. Ergosterol Peroxide Isolated from Ganoderma lucidum Abolishes MicroRNA miR-378-Mediated Tumor Cells on Chemoresistance. PLoS One 2012; 7:e44579; http://dx.doi.org/10.1371/journal.pone.0044579; PMID: 22952996
- LaPierre DP, Lee DY, Li SZ, Xie YZ, Zhong L, Sheng W, et al. The ability of versican to simultaneously cause apoptotic resistance and sensitivity. Cancer Res 2007; 67:4742 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-06-3610; PMID: 17510402
- Yee AJ, Akens M, Yang BL, Finkelstein J, Zheng PS, Deng Z, et al. The effect of versican G3 domain on local breast cancer invasiveness and bony metastasis. Breast Cancer Res 2007; 9:R47; http://dx.doi.org/10.1186/bcr1751; PMID: 17662123