Keywords: :
Although intensive regimens of antiretroviral therapy (HAART) reduce viral loads to undetectable levels in the circulation, HIV quickly resumes active replication when treatment is interrupted due to the emergence of the virus from latent reservoirs.Citation1,Citation2 Although it is difficult to exclude the possibility that slowly replicating viruses persist in sanctuary sites that are poorly accessed by the antiviral drugs, the consensus in the field is that the virus emerges from a small population of resting memory CD4 T cells (~1 in 106 cells) harboring silenced HIV proviruses.
Eliminating this latent reservoir is particularly challenging since it is established early during infection, is extremely stable (with an estimated half-life of 44 mo), and can be replenished during episodes of viremia or by homeostatic replacement of latently infected cells. Since latently infected cells express minimal levels of viral proteins they are invisible to the immune system and unaffected by antiretroviral drugs. Recent curative strategies have therefore focused on developing pharmaceutical agents that can induce HIV expression in latently infected cells and then purging these cells by antiviral immune responses, viral cytopathic effects or even cell-targeted killing strategies (the rhetorically named “shock and kill” strategy).Citation1,Citation2
In the last three months there has been a flurry of provocative papers, including the report by Boehm et al.Citation3 published in a previous issue of Cell Cycle, demonstrating that JQ1 and other clinically useful bromodomain (BET family) inhibitors can efficiently reverse HIV latency in established cell lines and in certain primary cell models for HIV latency.Citation4-Citation7 Thus the BET proteins have been validated as potential new targets for HIV induction strategies. But, how do these compounds work?
All BET proteins are highly conserved transcriptional regulators capable of binding to acetyl-lysine residues found on histones and many transcription factors through tandem bromodomains. Perhaps the best-studied BET family member is BRD4, which binds the positive transcription elongation factor b (P-TEFb) the essential cofactor for the HIV Tat gene (). It was therefore postulated that BET family inhibitors induce HIV expression by inhibiting BRD4 interactions with P-TEFb and thereby favoring enhanced Tat binding.Citation4,Citation6,Citation8 Consistent with this model, and the inhibitor studies, knockdown of BRD4 by shRNA also results in potent HIV induction.
Figure 1. Models for HIV induction by BET inhibitors. (A) Inhibition of BRD4 blocks its association with P-TEFb and permits enhanced association with the HIV transactivator protein Tat. The Tat:P-TEFb complex is recruited to the HIV promoter and induces transcription. (B) Inhibition of BRD2 blocks its association with E2F1:NFκB p50 heterodimers and co-repressor complexes. In the absence of BRD2, the repressor complexes are replaced by activator complexes, and HIV transcription is induced.
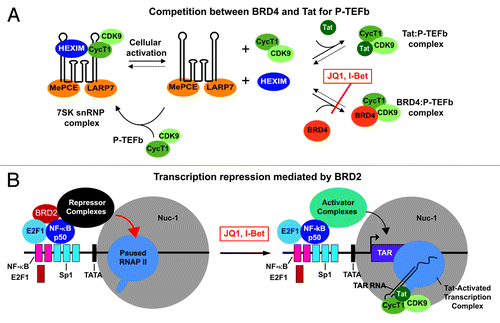
Boehm et al.Citation3 have now discovered that in addition to BRD4, a second BET protein, BRD2, also regulates HIV latency. Knockdown of BRD2 by shRNA activates HIV transcription to an even higher extent than knockdown of BRD4 and to levels comparable to JQ1 treatment of cells. In contrast to BRD4, BRD2 associates directly with transcription complexes and proteins required for chromatin remodeling. Thus it seems likely that BRD2 can enhance HIV transcription in response to JQ1 and other BET inhibitors. But how can it act as a repressor in the absence of BET inhibitors?
One clue comes from the observation that BRD2 interacts directly with the E2F1 transcription factor. Earlier studies showed that E2F1 can bind together with NFκB p50 to the HIV enhancer and block HIV transcription mediated by the NFκB p50/p65 heterodimer.Citation9 It seems reasonable to postulate that BRD2 is recruited to the HIV LTR by E2F1/p50 heterodimers, and then recruits repressor complexes carrying acetylated lysine residues (). Thus when BRD2 is inhibited, these interactions are blocked, and repressor complexes are exchanged for activators.
Although further work will be needed to confirm the molecular details of the two mechanisms outlined in , it is important to note at this stage that the high potency of JQ1 and related BET inhibitors in mediating HIV induction could be due to the targeting of multiple bromodomain proteins that regulate HIV transcription. Indeed, preliminary data emerging from several laboratories suggests that several other bromodomain proteins in addition to BRD4 and BRD2 can also contribute to the maintenance of HIV latency. It’s a safe bet that studies of these important regulatory mechanisms will reshape our understanding of HIV latency in the years to come.
Related Research Data
References
- Karn J. Curr Opin HIV AIDS 2011; 6:4 - 11; http://dx.doi.org/10.1097/COH.0b013e328340ffbb; PMID: 21242887
- Richman DD, et al. Science 2009; 323:1304 - 7; http://dx.doi.org/10.1126/science.1165706; PMID: 19265012
- Boehm D, et al. Cell Cycle 2012; 12; In press PMID: 23255218
- Li Z, et al. Nucleic Acids Res 2013; 41:277 - 87; http://dx.doi.org/10.1093/nar/gks976; PMID: 23087374
- Banerjee C, et al. J Leukoc Biol 2012; 92:1147 - 54; http://dx.doi.org/10.1189/jlb.0312165; PMID: 22802445
- Bartholomeeusen K, et al. J Biol Chem 2012; 287:36609 - 16; http://dx.doi.org/10.1074/jbc.M112.410746; PMID: 22952229
- Schröder S, et al. J Biol Chem 2012; 287:1090 - 9; http://dx.doi.org/10.1074/jbc.M111.282855; PMID: 22084242
- Zhu J, et al. Cell Rep 2012; 2:807 - 16; http://dx.doi.org/10.1016/j.celrep.2012.09.008; PMID: 23041316
- Kundu M, et al. J Biol Chem 1997; 272:29468 - 74; http://dx.doi.org/10.1074/jbc.272.47.29468; PMID: 9368006