Abstract
Human papilloma virus (HPV) infection represents an emerging risk factor in head and neck squamous cell carcinoma (HNSCC). In contrast to HPV-negative HNSCC, most cases of HPV-positive HNSCC encode wild-type p53, although the p53 protein in these cells is rapidly degraded via HPV E6-mediated ubiquitination and subsequent proteasomal degradation. This unique feature of HPV-positive HNSCC has raised hope that liberation of wild-type p53 from the E6 protein may have therapeutic benefit in this disease. Indeed, suppression of E6 expression promotes apoptosis in HPV-positive HNSCC cell lines. However, the role of p53 in mediating this cell death has not been determined. Here, we demonstrate that siRNAs targeting the E6/E7 RNA, or treatment with the proteasome inhibitor bortezomib, resulted in upregulation of functional p53 and p53 gene targets in three HPV-positive HNSCC cell lines, but not in HPV-negative HNSCC cells. Apoptosis induced by E6/E7 siRNA in HPV-positive cells was found to be dependent on p53, while bortezomib-induced cell death was modestly p53-dependent. Treatment with subtoxic doses of bortezomib led to cell cycle arrest in HPV-positive, but not HPV-negative HNSCC cells. Moreover, this cell cycle arrest was mediated by p53 and the cell cycle inhibitor p21, the product of a p53 target gene. Collectively, these findings establish that wild-type p53 encoded by HPV-positive HNSCC cells, once liberated from HPV E6, can play important roles in promoting apoptosis and cell cycle arrest.
Introduction
Head and neck cancer refers to malignancies of the oral cavity, larynx and pharynx, and is the sixth most common cancer worldwide.Citation1 More than 90% of these cancers are squamous cell carcinomas and are referred to as head and neck squamous cell carcinomas (HNSCCs). Unfortunately, progress has been slow in identifying effective therapeutic strategies for HNSCC, and 5-y survival rates have remained around 50% for the past few decades, underscoring the need to develop new approaches.Citation2 Known risk factors for HNSCC include alcohol and tobacco consumption and infection with human papilloma virus (HPV).Citation3-Citation7 HPV represents an emerging health issue in HNSCC. While the overall incidence of HNSCC is slowly declining in many parts of the world, the incidence of HPV-positive disease is increasing, particularly among younger adults in the United States and Europe.Citation8-Citation11
Several interesting features characterize HPV-positive HNSCC. First, most HPV-positive HNSCCs derive from the oropharynx, where roughly 50% of cases may harbor the virus.Citation2,Citation7,Citation12 Second, patients with HPV-positive HNSCC generally respond better to therapy and have better prognoses than HPV-negative patients.Citation13-Citation19 Third, in contrast to HPV-negative HNSCC where p53 and RB are almost always mutated or missing, most HPV-positive HNSCCs encode wild-type p53 and RB.Citation20-Citation22 Mutation or deletion of p53 and RB may be unnecessary in HPV-positive HNSCC, since the wild-type p53 and RB produced in these cells are routinely degraded by HPV E6 and E7 proteins, respectively, which promote their ubiquitination and proteasomal degradation.Citation23,Citation24 Despite the continual degradation of p53 and RB in HPV-positive HNSCC, presumptions have been made that the better therapeutic responses of these cancers is due to the potential to produce the wild-type 53 and RB proteins. However, this hypothesis has never been directly tested.
Considerably more has been learned about the role of HPV in carcinogenesis from studies of cervical cancer, where nearly all cases can be attributed to HPV infection. Integration of high-risk HPVs into genomic DNA attenuates expression of the HPV E2 protein, a transcriptional repressor of the viral E6 and E7 genes, which are transcribed as a bicistronic RNA.Citation25 Similar to HPV-positive HNSCC, the p53 and RB proteins produced in cervical cancer cells are wild-type proteins, but are degraded by the actions of the upregulated E6/E7 proteins. Enforced expression of bovine or human papilloma virus E2 protein in cervical cancer cells has been shown to suppress E6/E7 expression and induce apoptosis, growth arrest and senescence.Citation26-Citation31 Highly similar effects are seen following suppression of E6/E7 using siRNA or shRNA strategies.Citation32-Citation34 Downregulation of E6 in cervical cancer cells is accompanied by upregulation of wild-type p53 and expression of p53 target genes.Citation27,Citation28,Citation31 Importantly, inhibition of the upregulated p53 through expression of dominant-negative p53 has indicated roles for the wild-type protein in cervical cancer growth arrest, apoptosis and senescence.Citation27,Citation35 In the case of HPV-positive HNSCC, siRNA/shRNA suppression of E6 expression in two cell lines has been shown to induce p53 and activate apoptosis.Citation36,Citation37 It is important to determine the role of p53 in this scenario, since E6 is known to bind to multiple other proteins, and the consequences of suppressing E6 may be due to disruption of these p53-independent interactions/pathways. However, due to a lack of functional testing, the contributions and roles of wild-type 53 in mediating either apoptosis or growth arrest in HPV-positive HNSCC have not been determined.
In this report we used three HPV-positive and three HPV-negative HNSCC cell line models to investigate the role of p53. Suppression of E6/E7 expression using siRNAs led to upregulation of wild-type p53 protein in HPV-positive cells but not HPV-negative cells. Although peptide and small-molecule inhibitors targeting E6 have recently been identified, they are still in early development and face significant hurdles prior to translation to the clinic.Citation38-Citation43 Therefore, as an alternative approach for inhibiting E6 activity, we employed treatment with bortezomib, a Food and Drug Administration-approved proteasome inhibitor. Similar to treatment with E6/E7 siRNAs, bortezomib induced expression of wild-type p53 in HPV-positive cells, but not HPV-negative cells. Inhibition of p53 expression attenuated apoptosis caused by E6/E7 siRNAs, and modestly inhibited cell death resulting from bortezomib treatment. Notably, subtoxic levels of bortezomib induced cell cycle arrest in HPV-positive HNSCC cells, without impacting cell cycle progression in HPV-negative cells. Bortezomib-induced cell cycle arrest was found to be dependent on p53, as well as the downstream mediator p21. These findings establish important roles for wild-type p53 in HPV-positive HNSCC, and suggest that liberation of p53 from negative regulation by E6 may have therapeutic value in the treatment of this disease.
Results
Expression status of HPV16 E6/E7 RNA in HNSCC cell line models
Our studies incorporated three HNSCC cell lines that have been reported to be HPV16-positive (UPCI:SCC090, UM-SCC-47, UD-SCC-2) and three HNSCC cell lines that are HPV-negative (UM-SCC-22A, UM-SCC-1, 1483).Citation44 To verify the expression status of HPV16 E6/E7 bicistronic RNA in our models, RT-PCR was performed using primers pairs specific for E6 coding sequences or E7 coding sequences (). Use of the E6 primers yielded a predicted 298 bp fragment with RNA from UPCI:SCC090, UM-SCC-47 and UD-SCC-2 cells. Similarly, a predicted 315 bp fragment was seen using the E7 primers. By contrast, RT-PCR failed to generate E6 and E7 bands using RNA from UM-SCC-22A, UM-SCC-1 and 1483 cells, despite successful generation of a control β-actin fragment. These experiments confirmed the expression status of E6/E7 RNA in the HNSCC cell lines.
Figure 1. Detection of HPV E6/E7 in HNSCC cell lines. (A) RT-PCR was performed on total RNA harvested from HNSCC cell lines as described in Materials and Methods. Expected sizes of PCR fragments were detected using E6-specific primers (298 bp) and E7-specific primers (315 bp) in UPCI:SCC090, UM-SCC-47 and UD-SCC-2 cells. Expression of β-actin RNA was used as a control. (B) Three HPV-positive HNSCC cell lines were transfected with 100 nM nonspecific siRNA or siRNA directed against the E6/E7 bicistronic RNA. After 48 h, total RNA was prepared and E6/E7 RNA expression was assessed by RT-PCR. Similar results were obtained in three independent experiments.
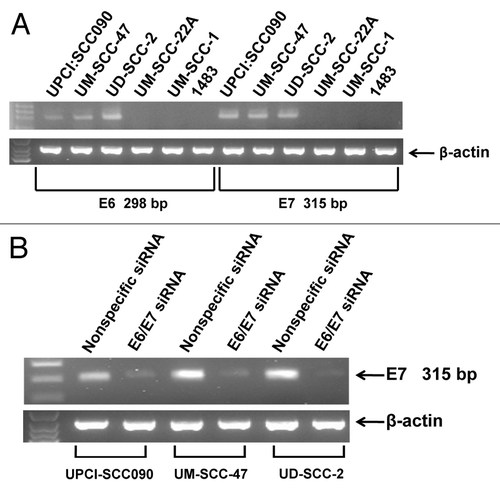
To validate the specificity of the RT-PCR-based expression analysis, and to achieve downregulation of E6/E7 expression, the three HPV-positive cell lines were transfected with a nonspecific siRNA or an siRNA targeting the E6/E7 RNA. Forty-eight h after transfection, RT-PCR was performed using the E6 and E7 primer pairs (). Transfection with E6/E7 siRNA led to marked downregulation of the 315 bp E7 fragment () and 298 bp E6 fragment (not shown), relative to transfection with nonspecific siRNA. This confirmed the identities of the RT-PCR fragments and the utility of the E6/E7 siRNA.
Suppression of E6/E7 expression induces p53 and activates p53-dependent apoptosis signaling
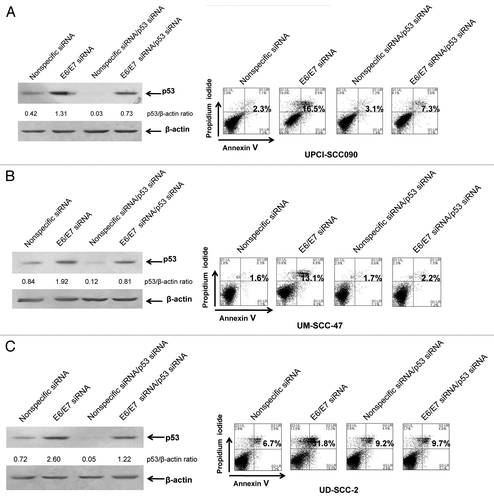
The E6 protein is known to promote ubiquitination and proteasomal degradation of p53.Citation23 In cervical cancer cells, suppression of E6 expression results in p53 upregulation and activation of p53-dependent apoptosis.Citation27,Citation35 Suppression of E6 expression using an siRNA approach has also been shown to upregulate p53 in HPV-positive HNSCC cells, but the role and impact of this upregulated p53 on subsequent cellular responses has not been determined.Citation36,Citation37 As depicted in (left panels), transfection of E6/E7 siRNA led to a 3.1-, 2.3- and 3.6-fold induction of p53 protein in UPCI:SCC090, UM-SCC-47 and UD-SCC-2, respectively, when compared with transfection with nonspecific siRNA. Flow cytometric analysis of Annexin V/PI staining (, right panels) also demonstrated induction of apoptosis 72 h following transfection of E6/E7 siRNA, consistent with similar findings by Rampias et al.Citation37 To determine the role of p53 in apoptosis induction by E6/E7 siRNA, the HPV-positive cell lines were co-transfected with E6/E7 siRNA and p53 siRNA (). Immunoblotting revealed that co-transfection with p53 siRNA inhibited by approximately 50% the upregulation of p53 resulting from E6/E7 siRNA. Importantly, suppression of p53 expression inhibited apoptosis induction by E6/E7 siRNA, as assessed by Annexin V staining. These results demonstrate that the apoptosis activated when E6/E7 expression is inhibited in HPV-positive HNSCC cells is p53-dependent.
Figure 2. E6/E7 siRNA induces p53-dependent apoptosis in HPV-positive HNSCC cells. UPCI:SCC090 (A), UM-SCC-47 (B) and UD-SCC-2 (C) cells were transfected with 100 nM nonspecific or E6/E7 siRNAs alone (left, lanes 1 and 2; right, panels 1 and 2), or at 50 nM in combination with 50 nM p53 siRNA (left, lanes 3 and 4; right, panels 3 and 4). After 72 h, cells were harvested and subjected to immunoblotting for p53 and β-actin (loading control) or flow cytometric analysis of Annexin V/PI staining. Numbers on the immunoblots indicate p53/β-actin ratios, as determined by densitometric analysis. Suppression of E6/E7 led to induction of p53, which was partially prevented by p53 siRNA. Numbers on flow diagrams indicate the percentage of Annexin V-positive cells. Apoptosis induced by E6/E7 suppression was dependent on p53.
Bortezomib induces functional p53 in HPV-positive HNSCC cells
The ability of p53 to mediate apoptosis activated by E6/E7 siRNA in HPV-positive cells indicated that the p53 expressed by these cells is functional. This is consistent with observations that most HPV-positive HNSCC tumors encode wild-type p53.Citation19,Citation21,Citation22 Our results also underscore the potential benefit of liberating p53 from E6 negative regulation in HPV-positive HNSCC. Since E6 is known to promote proteasomal degradation of p53, we, therefore, evaluated proteasome inhibition as an alternative mechanism for liberating wild-type p53 in HPV-positive HNSCC cells.
In the absence of proteasome inhibitor, immunoblotting () revealed high levels of p53 expression in two HPV-negative cell lines (UM-SCC-22A and 1483) and lack of p53 in the other HPV-negative line (UM-SCC-1). However, expression of p21 was not detected in any of the HPV-negative lines, suggesting that the abundant p53 detected in UM-SCC-22A and 1483 is mutant/nonfunctional (). Following treatment of the HPV-negative lines with the proteasome inhibitor bortezomib, no significant increases in p53 or p21 levels were detected (). By contrast, treatment with bortezomib led to substantial upregulation of p53 in all three HPV-positive cell lines (). Moreover, bortezomib upregulation of p53 in the HPV-positive cells was accompanied by upregulation of p21, indicating that the induced p53 in these cell lines is functional (). Interestingly, determination of IC50 values for bortezomib revealed similar ranges of sensitivity in HPV-negative (15.2–51.3 nM) and HPV-positive (10.0–110.4 nM) cell lines ().
Figure 3. Proteasome inhibition with bortezomib induces functional p53 protein in HPV-positive cells but not HPV-negative cells. Three HPV-negative HNSCC cell lines (A) and three HPV-positive HNSCC cell lines (B) were left untreated, treated for 24 h with 0.1% DMSO, or treated for 24 or 48 h with 50 nM (UM-SCC-22A, UM-SCC-1, 1483, UPCI:SCC090, UM-SCC-47) or 200 nM (UD-SCC-2) bortezomib. Following treatment, immunoblotting was used to detect expression of p53, p21 and β-actin. Ratios of p53/β-actin and p21/β-actin were determined by densitometric scanning. Similar results were obtained in three independent experiments. (C) Suppression of p53 expression/activity in bortezomib-treated HPV-positive cells using p53 siRNA. HPV-positive HNSCC cells were either left untransfected or were transfected with 100 nM nonspecific siRNA or p53 siRNA. After 24 h, untransfected cells were treated with 0.1% DMSO, 50 nM (UPCI:SCC090, UM-SCC-47) or 200 nM (UD-SCC-2) bortezomib, and transfected cells were treated with bortezomib (50 or 200 nM, as above). After 48 h of treatment, immunoblotting was used to detect p53, p21, HDM2 and β-actin. Bortezomib induction of p53 and the products of p53 target genes were inhibited by p53 siRNA, but not nonspecific siRNA. The experiment was performed three times with similar results.

Table 1. HNSCC cell line characteristics
Bortezomib-induced cell death is partially dependent on p53 in HPV-positive, but not HPV-negative, cells
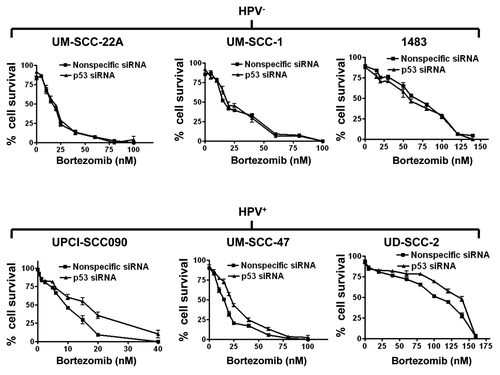
We next sought to determine the impact of p53 on bortezomib-induced HNSCC cell death. Transfection with p53 siRNA was used to achieve downregulation of p53. As shown in , transfection with p53 siRNA abolished bortezomib-induced upregulation of p53 in the three HPV-positive cell lines. Similarly, p53 siRNA abolished expression of the p53 targets, p21 and HDM2 (). Transfection with nonspecific siRNA, on the other hand, had little, if any, effect on bortezomib induction of p53, p21 and HDM2 in the HPV-positive cell lines. We then determined the impact of p53 downregulation on sensitivity to bortezomib in trypan blue exclusion assays (). As expected, transfection with p53 siRNA had no observable effect on bortezomib-induced cell death in the HPV-negative cell lines. In the HPV-positive cell lines, however, downregulation of p53 resulted in moderately enhanced resistance to bortezomib. These results indicate that liberation of functional p53 provides a modest contribution to bortezomib-induced cell death in HPV-positive HNSCC cells.
Figure 4. Bortezomib-induced cell death is modestly dependent on p53 in HPV-positive cells, but independent of p53 in HPV-negative cells. HPV-negative (top panels) and HPV-positive (bottom panels) HNSCC cells were transfected with 100 nM nonspecific siRNA or p53 siRNA. Twenty-four h after transfection, cells were treated for 48 h with varying concentrations of bortezomib, followed by assessment of cell viabilities by trypan blue exclusion. Error bars represent the standard deviations of triplicate wells. The experiment was performed three times with similar results.
Subtoxic doses of bortezomib inhibit growth and induce cell cycle arrest in HPV-positive, but not HPV-negative, HNSCC cells
We next evaluated the impact of bortezomib-induced p53 on cellular proliferation and cell cycle progression. For these experiments, it was necessary to utilize a subtoxic dose of bortezomib. Preliminary experiments determined that 5 nM bortezomib was lower than the IC50 for all six cell lines (), failed to induce appreciable cell death, yet still was capable of upregulating p53 protein in HPV-positive cells. HPV-negative and HPV-positive cell lines were treated with 5 nM bortezomib for varying lengths of time, followed by determination of viable cell numbers (). As control, cells were treated with equivalent concentrations of DMSO (0.1%), the drug diluent. After 8 d of treatment, bortezomib did not significantly impact the proliferation of any of the three HPV-negative cell lines. By contrast, bortezomib significantly inhibited proliferation of all three HPV-positive lines. In particular, very potent inhibition was seen in UPCI:SCC090 and UM-SCC-47 cells. Although less inhibition was seen in UD-SCC-2 cells, it is notable that these cells exhibited the highest IC50 (110.4 nM; ) of all six cell lines, hinting that there may be other mechanisms of bortezomib resistance operating in these cells.
Figure 5. Low-dose bortezomib inhibits proliferation and induces cell cycle arrest in HPV-positive, but not HPV-negative, HNSCC cells. (A) HPV-negative (top panels) and HPV-positive (bottom panels) cells were plated in triplicate in 96-well plates, allowed to recover overnight, then treated for varying lengths of time with either 0.1% DMSO or 5 nM bortezomib. Following treatment, viable cell numbers were determined in trypan blue exclusion assays. p values were determined by comparing DMSO- vs. bortezomib-treated cells on days 5–8. (B) HPV-negative (top panels) and HPV-positive (bottom panels) cells were treated for 48 h with 0.1% DMSO or 5 nM bortezomib, followed by flow cytometric determination of cell cycle status, as described in Materials and Methods. Columns represent the means from three independent experiments and error bars represent the standard error of the means.
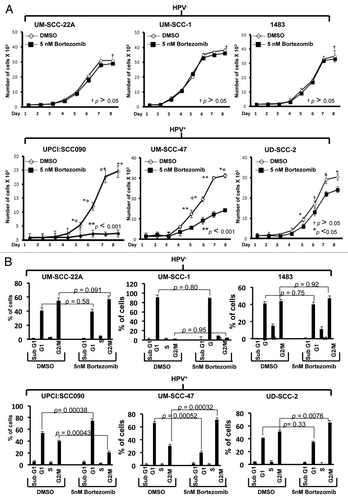
The ability of subtoxic bortezomib to inhibit proliferation in HPV-positive, but not HPV-negative cell lines suggested potential effects on cell cycle. Therefore, cells were treated with 5 nM bortezomib followed by assessment of cell cycle status by flow cytometry (). In the HPV-negative cells, no significant effects on cell cycle were detected. In HPV-positive cells, however, treatment with bortezomib resulted in G1 accumulation in UPCI:SCC090 cells and G2/M accumulation in UM-SCC-47 and UD-SCC-2 cells. Although it is unclear why G1 arrest was observed in one HPV-positive cell line and G2/M in the other two, the ability of bortezomib to induce both G1 and G2/M arrest has been reported in other malignancies.Citation45-Citation47
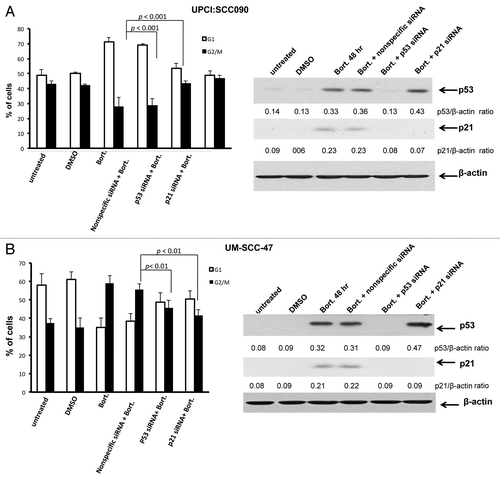
Bortezomib-induced cell cycle arrest in HPV-positive HNSCC cells is mediated by p53 and p21
The p53 protein, acting primarily through p21, is known to promote cell cycle arrest at G1 and G2/M. Therefore, we sought to determine whether the bortezomib-induced cell cycle arrest observed in HPV-positive cells was mediated by the liberated, functional p53 expressed in these cells. Cells were treated with bortezomib, leading to G1 arrest in UPCI:SCC090 cells () and G2/M arrest in UM-SCC-47 cells (). Prior transfection with nonspecific siRNA did not significantly impact these effects of bortezomib on the cell cycle, nor the induction of p53 and p21 proteins (). On the other hand, transfection with p53 siRNA abolished bortezomib induction of both p53 and p21 and inhibited G1 arrest in UPCI:SCC090 cells and G2/M arrest in UM-SCC-47 cells. Transfection with p21 siRNA abrogated p21 induction, without impacting p53 upregulation. Moreover, suppression of p21 alone inhibited G1 and G2/M arrest to the same degree as was achieved by suppression of p53 expression. These results demonstrate that the wild-type p53 liberated by proteasome inhibition in HPV-positive cells mediates the subsequent cell cycle arrest. In addition, the effects of p53 on cell cycle arrest in the HPV-positive cells are dependent on p21.
Figure 6. Bortezomib-induced cell cycle arrest in HPV-positive HNSCC cells is mediated by p53 and p21. HPV-positive UPCI:SCC090 (A) and UM-SCC-47 (B) cells were mock transfected, or were transfected with nonspecific siRNA, p53 siRNA, or p21 siRNA. The transfected cells were incubated for 24 h before any subsequent treatment. The mock transfected cells were then either left untreated, or were treated for 48 h with 0.1% DMSO or 5 nM bortezomib. The transfected cells were treated for 48 h with 5 nM bortezomib. Following treatment, cell cycle status was determined by flow cytometry (left panels). Columns represent the means of three independent experiments, and error bars the standard error of the means. Immunoblotting (right panels) was used to demonstrate inhibition of p53 expression by p53 siRNA, and inhibition of p21 expression by p53 or p21 siRNAs. Immunoblotting was performed on each of the three independent experiments analyzed by flow cytometry, with similar results obtained.
Discussion
Multiple clinical studies have determined that HPV-positive HNSCC patients typically respond better to chemoradiation therapy and have better prognoses than patients with HPV-negative HNSCC.Citation13-Citation18 This has raised hope that it may be possible to target unique elements of HPV-positive HNSCC to achieve even better outcomes in this emerging disease. One such unique feature is the fact that HPV-positive HNSCCs, in contrast to HPV-negative HNSCCs, generally encode wild-type p53 and RB, which are continually degraded via the HPV E6 and E7 proteins, respectively. Studies by Ferris et al.Citation36 and Rampias et al.Citation37 have shown that siRNA/shRNA-mediated downregulation of E6/E7 RNA in HPV-positive HNSCC cell lines results in p53 upregulation, while also inducing apoptosis. However, the potential dependence of this apoptosis on p53 was not investigated. Therefore, while these studies established the value of targeting E6/E7 RNA in HPV-positive HNSCC, the benefit and impact of liberating wild-type p53 remained unclear. Here, we used two distinct approaches to liberate p53 in a panel of HPV-positive HNSCC cell lines: siRNA suppression of E6/E7 RNA and proteasome inhibition with bortezomib. Moreover, we used siRNA to prevent p53 upregulation caused by these approaches, allowing us to evaluate the role and impact of p53 on apoptosis and cell cycle arrest in the HPV-positive HNSCC cells. Our findings demonstrate upregulation of p53 and the p53 targets p21 and HDM2 following E6/E7 suppression or proteasome inhibition. Cell death resulting from E6/E7 siRNAs or treatment with bortezomib was inhibited by preventing p53 expression. A role for p53 in promoting cell cycle arrest in HPV-positive HNSCC cells was also observed. Treatment with subtoxic doses of bortezomib resulted in p53- and p21-dependent cell cycle arrest in HPV-positive, but not HPV-negative, HNSCC cells. Collectively, these findings demonstrate that wild-type p53 liberated from E6 negative regulation mediates induction of cell death and cell cycle arrest in HPV-positive HNSCC, and point to the potential value of therapeutic strategies that facilitate liberation of p53 in this disease.
The necessity of developing new therapeutic strategies and reagents in HNSCC is highlighted by two points. First, the success rates for chemoradiation in treating advanced HNSCC, including HPV-positive HNSCC, have improved only incrementally over the past few decades, and this remains a difficult to treat and deadly disease.Citation2 Second, the incidence of HPV-positive HNSCC continues to increase in the United States and several European nations.Citation8-Citation11 In the case of HPV-positive cervical cancer, the causative role of infection with high-risk HPVs is well-established. This has led to the development and application of HPV vaccines as highly effective tools for preventing cervical cancer. The causative role of HPV infection in certain HNSCCs is an area of more recent investigation. Epidemiologic evidence has noted a strong correlation between HPV infection and tumors of the oropharynx.Citation2,Citation7,Citation12 As confirmed in this report, suppression of E6/E7 expression leads to induction of cell death in HPV-positive HNSCC cell lines. Moreover, co-expression of E6 and E7 under the direction of the keratin 14 gene promoter leads to synergistic development of head and neck tumors in a transgenic mouse model, although E7 is more dominant than E6 when expressed individually.Citation48,Citation49 Thus, it is possible that vaccination against high-risk HPVs also may have significant impact on the development of HPV-positive HNSCC. However, studies validating anti-HPV vaccines as effective agents against HNSCC development have not been reported and may take years to accumulate statistically significant results.
Alternative approaches toward treating HPV-positive HNSCC may take advantage of the unique characteristics of this disease. HPV-positive HNSCC is now considered a distinct disease entity from tobacco-induced HNSCC.Citation12 Further, HPV-positive HNSCC patients typically exhibit better responses to chemoradiation and have better clinical prognoses than HPV-negative patients. An obvious molecular distinction of HPV-positive HNSCC is the continuous expression of HPV E6 and E7 proteins in the tumor cells. Our results and those of others demonstrate the utility of suppressing E6/E7 RNA expression in vitro.Citation36,Citation37 In vivo suppression of E6/E7 has been achieved in cervical cancer. Fujii et al.Citation50 have shown that intratumoral injection of siRNA targeting HPV18 E6/E7 RNA inhibited the growth of xenograft tumors derived from SKG-II cervical cancer cells. Additionally, Gu et al.Citation51 demonstrated that systemic delivery of lentiviral HPV18 E6/E7 shRNA yielded antitumor effects on HeLa cell (cervical cancer line) xenograft tumors. It seems likely that in vivo administration of E6/E7 siRNA/shRNA will result in similar effects on HPV-positive HNSCC xenograft tumors, although this remains to be tested.
Although suppression of E6/E7 expression represents a viable approach against HPV-positive HNSCC, the mechanism whereby E6/E7 suppression leads to induction of HNSCC cell death has remained unclear. Our results establish a clear role for liberation of wild-type p53 in promoting the death of these cells. However, in vivo application of E6/E7 siRNAs/shRNAs as therapeutic agents may be hindered by several factors. For this reason we investigated an alternative approach for liberating wild-type p53: inhibition of E6-mediated ubiquitination and proteasomal degradation of the p53 protein. This was achieved via inhibition of the proteasome with bortezomib, a compound that is already approved by the Food and Drug Administration for the treatment of multiple myeloma and mantle cell lymphoma.Citation52-Citation55 As expected, bortezomib treatment resulted in upregulation of functional p53 protein in HPV-positive HNSCC cells, but not in HPV-negative HNSCC cells. Inhibition of p53 upregulation resulted in modest inhibition of bortezomib-induced cell death, indicating an anticancer effect for liberation of p53 by proteasome inhibition. The minor impact of p53 on bortezomib-induced cell death suggests that this agent induces apoptosis via p53-independent pathways as well. In addition to a role in promoting cell death, we also discovered that p53 liberated from E6 mediated cell cycle arrest in HPV-positive HNSCC cells treated with subtoxic doses of bortezomib. Taking advantage of this HPV-specific mechanism may have therapeutic benefit for treatment of HPV-positive disease. In this regard, Pyeon et al.Citation56 have performed genome-wide expression profiling in HNSCC and cervical cancers. They observed similarities in deregulated expression of cell cycle genes in HPV-positive HNSCCs and HPV-positive cervical cancers, which were distinct from those observed in HPV-negative HNSCCs. Further studies will be needed to determine whether the wild-type p53 encoded by these HPV-positive cancers contributes to the altered expression of key cell cycle regulators beyond p21. p53 is also known to promote upregulation of BAX, PUMA, NOXA and BID, pro-apoptotic members of the Bcl-2 protein family,Citation57-Citation61 which may contribute to the death-inducing activities of p53 in HPV-positive disease. In addition, p53 has been shown to associate with pro-apoptotic BAX and BAK, resulting in their activation,Citation62-Citation64 and also with Bcl-2 and Bcl-XL, resulting in inhibition of these anti-apoptotic proteins.Citation63,Citation65 Whether these mechanisms are important for the anticancer effects of wild-type p53 in HPV-positive HNSCC remains to be determined.
In summary, our studies have established roles for wild-type p53 encoded by HPV-positive HNSCC in mediating and promoting cell cycle arrest and apoptosis. These findings support the development and testing of therapeutic strategies aimed at releasing p53 in HPV-positive HNSCC from negative regulation by E6. Further studies are needed to evaluate the impact of liberating RB from E7 in this disease. In addition, future studies can now be more carefully considered to determine whether liberation of wild-type p53 or RB in HPV-positive HNSCC patients accounts for the improved responsiveness of these patients to conventional therapies.
Materials and Methods
Cells and reagents
Three HPV-negative HNSCC cell lines (UM-SCC-22A, UM-SCC-1, 1483) and three HPV-positive HNSCC cell lines (UPCI:SCC090, UM-SCC-47, UD-SCC-2) were studied. UM-SCC-22A, UM-SCC-1 and UM-SCC-47 were kindly provided by Dr. Thomas Carey (University of Michigan) and 1483 were provided by Dr. Gary Clayman (MD Anderson Cancer Center). UPCI:SCC090 were given by Susanne Gollin (University of Pittsburgh) and UD-SCC-2 by Kathrin Scheckenbach (University of Duesseldorf). All cell lines were cultured in DMEM medium containing 10% fetal bovine serum supplemented with 100 units/ml of penicillin/streptomycin. Genotypic validation of the cell lines was performed using the AmpFISTR Profiler Plus Kite (PE Biosystems).
Antibodies directed against p53 (cat. #sc-55476), p21(#sc-6246) and HDM2 (#sc-13161) were purchased from Santa Cruz Biotechnology, Inc. Anti-β-actin was from Sigma (#A5441), and horse-radish peroxidase-conjugated secondary antibodies were from Promega (#W4028 and #W4018). Bortezomb was obtained from LC Laboratories (#B-1408). Annexin V/propidium iodide apoptosis detection kits were purchased from BD BioScience, Inc. (cat. #556547).
Detection of E6/E7 RNA
For detection of E6/E7 RNA encoded by HPV, total RNA was prepared from 2 million cells using TRIzol Reagent (Life Technologies Corp.; #15596-018) extraction. One µg of total RNA was converted to cDNA then subjected to PCR amplification using SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen, #12574-018), according to the manufacturer’s instructions. The following primers were used: E6 forward primer: ‘5-TTACCACAGTTATGCACAGA-3′; E6 reverse primer: ‘5-ACAGTGGCTTTTGACAGTTA-3′; E7 forward primer: ‘5-AGAAACCCAGCTGTAATCAT-3′; E7 reverse primer: ‘5-TTATGGTTTCTGAGAACAGA-3′; β-actin forward primer: ‘5-AGCCATGTACGTTGCTATCC-3′; β-actin reverse primer: ‘5-TTGGCGTACAGGTCTTTGC-3′. The RT-PCR products were electrophoresed on 2% agarose gels, stained with ethidium bromide and photographed using Molecular Imager ChemiDoc XRS+ with Image Lab software (Bio-Rad Laboratories).
Immunoblotting
Treated cells (3 × 106) were harvested by cell scraping, washed once in cold PBS and lysed in 300 μl of lysis buffer (50 mM Tris pH = 8.0, 150 mM NaCl, 0.1% SDS, 1% NP40) containing one tablet of Protease Inhibitor Cocktail (Roche Applied Sciences; #11836153001)/10 mls. Lysates were subjected to sonication for 6 sec, then clarified by microcentrifugation at 13,000 rpm for 30 min at 4°C. Protein concentrations were determined using Bio-Rad Protein Assay dye concentrate (Bio-Rad Laboratories, Inc.; #500-0006). Equal quantities of protein lysate (40 µg/lane) were electrophoresed on 12.5% SDS-PAGE gels, then transferred to nitrocellulose filters. Filters were blocked for 1 h at room temperature with 5% fat-free milk prepared in TBST (10 mM Tris pH = 8.0, 150 mM NaCl, 0.1% Tween 20), rinsed once in TBST and incubated overnight at 4°C with primary antibodies in 5% fat-free milk prepared in TBST. After rinsing three times in TBST, filters were incubated with secondary antibodies for 1 h at room temperature in TBST containing 1% fat-free milk. Filters were then rinsed again in TBST and developed using Western Lightning Chemiluminescence Reagent Plus (Perkin-Elmer Life Science; #NEL 102001EA).
Knockdown of p53, p21 and E6/E7
siRNAs targeting p53 and p21 RNAs, as well as the nonspecific control siRNA, were obtained from Santa Cruz Biotechnology, Inc. (#sc-29437, #sc-44214 and # sc-37007, respectively). siRNAs for knocking down E6/E7 (5′-GUAUGGAACAACAUUAGAA-3′) were synthesized by Ambion, Inc. For transfection of the siRNAs, 0.3 × 106 UM-SCC-47, 0.35 × 106 UPCI:SCC090, or 0.4 × 106 of UD-SCC-2 were plated into 60-mm plates and allowed to grow for 24 h. The cells were then individually transfected with 100 nM of siRNA using Lipofectamine 2000 (Invitrogen; #11668-019), as previously described.Citation66 Twenty-four h after transfection the cells were subjected to treatment with vehicle or bortezomib.
Assays of cell viability and apoptosis
Cell viabilities were determined by trypan blue exclusion assays. For determination of bortezomib IC50 values, cells were first plated in triplicate into 96-well plates (3,000 cells/well for UM-SCC-1 and UM-SCC-47; 4,000 cells/well for 1483 and UPCI:SCC090; 6,000 cells/well for UM-SCC-22A and UD-SCC-2), so that the cells were roughly 40~50% confluent. After overnight recovery, cells were treated with varying concentrations of bortezomib for 48 h. Floating cells were then combined with cells detached from the plate by trypsinization, then treated with trypan blue. A minimum of 200 cells were counted for each data point, and IC50 values were calculated using GraphPad Prism software (GraphPad Software, Inc.). Activation of apoptosis signaling was assessed by Annexin V/propidium iodide (PI) staining using FITC Annexin V Apoptosis Detection kits (BD PharMingen; #556547). Floating cells were combined with cells detached by trypsinization, then subjected to microcentrifugation, washing in cold PBS and resuspension in Annexin V/PI binding buffer. The cells were then stained according to the manufacturer’s instructions. The stained cells were analyzed by flow cytometry using a BD Accuri C6 flow cytometer (BD Biosciences).
Cell cycle analysis
Cells were plated into 6-well plates (0.2 × 106/well for UM-SCC-47 and UM-SCC-1; 0.3 × 106/well for UPCI:SCC090, UM-SCC-22A and 1483; 0.5 × 106/well for UD-SCC-2), allowed to recover overnight, then treated for 48 h. Following treatment, floating cells were combined with attached cells, then subjected to centrifugation at 3,000 rpm for 5 min at 4°C. The cell pellets were washed with cold PBS, then fixed for a minimum of 1 h with 70% ethanol at 4°C. The fixed cells were washed with AnnexinV/PI binding buffer, stained with PI (10 µg/ml) and analyzed by flow cytometry.
Statistics
Data was analyzed using SigmaStat software (Jandel Scientific Software). For comparisons of two groups we applied Student’s t-test. For comparions of multiple groups we used one-way ANOVA followed by Student’s Newman-Keul’s tests. p values < 0.05 were considered statistically significant.
Acknowledgments
We thank Drs Robert L. Ferris, Jennifer R. Grandis and Yan Zang for valuable discussions and for providing reagents. This work was supported by NIH grants R01 CA137260 and P50 CA097190. This project also used UPCI shared resources supported in part by P30CA047904.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225 - 49; http://dx.doi.org/10.3322/caac.20006; PMID: 19474385
- Fung C, Grandis JR. Emerging drugs to treat squamous cell carcinomas of the head and neck. Expert Opin Emerg Drugs 2010; 15:355 - 73; http://dx.doi.org/10.1517/14728214.2010.497754; PMID: 20557270
- Hashibe M, Brennan P, Benhamou S, Castellsague X, Chen C, Curado MP, et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst 2007; 99:777 - 89; http://dx.doi.org/10.1093/jnci/djk179; PMID: 17505073
- Hashibe M, Brennan P, Chuang SC, Boccia S, Castellsague X, Chen C, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev 2009; 18:541 - 50; http://dx.doi.org/10.1158/1055-9965.EPI-08-0347; PMID: 19190158
- Mork J, Lie AK, Glattre E, Hallmans G, Jellum E, Koskela P, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med 2001; 344:1125 - 31; http://dx.doi.org/10.1056/NEJM200104123441503; PMID: 11297703
- Herrero R, Castellsagué X, Pawlita M, Lissowska J, Kee F, Balaram P, et al, IARC Multicenter Oral Cancer Study Group. Human papillomavirus and oral cancer: the International Agency for Research on Cancer multicenter study. J Natl Cancer Inst 2003; 95:1772 - 83; http://dx.doi.org/10.1093/jnci/djg107; PMID: 14652239
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 2000; 92:709 - 20; http://dx.doi.org/10.1093/jnci/92.9.709; PMID: 10793107
- Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the U.S. population ages 20-44 years. Cancer 2005; 103:1843 - 9; http://dx.doi.org/10.1002/cncr.20998; PMID: 15772957
- Näsman A, Attner P, Hammarstedt L, Du J, Eriksson M, Giraud G, et al. Incidence of human papillomavirus (HPV) positive tonsillar carcinoma in Stockholm, Sweden: an epidemic of viral-induced carcinoma?. Int J Cancer 2009; 125:362 - 6; http://dx.doi.org/10.1002/ijc.24339; PMID: 19330833
- Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol 2008; 26:612 - 9; http://dx.doi.org/10.1200/JCO.2007.14.1713; PMID: 18235120
- Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011; 29:4294 - 301; http://dx.doi.org/10.1200/JCO.2011.36.4596; PMID: 21969503
- Gillison ML. Human papillomavirus-associated head and neck cancer is a distinct epidemiologic, clinical, and molecular entity. Semin Oncol 2004; 31:744 - 54; http://dx.doi.org/10.1053/j.seminoncol.2004.09.011; PMID: 15599852
- Lindel K, Beer KT, Laissue J, Greiner RH, Aebersold DM. Human papillomavirus positive squamous cell carcinoma of the oropharynx: a radiosensitive subgroup of head and neck carcinoma. Cancer 2001; 92:805 - 13; http://dx.doi.org/10.1002/1097-0142(20010815)92:4<805::AID-CNCR1386>3.0.CO;2-9; PMID: 11550151
- Li W, Thompson CH, O’Brien CJ, McNeil EB, Scolyer RA, Cossart YE, et al. Human papillomavirus positivity predicts favourable outcome for squamous carcinoma of the tonsil. Int J Cancer 2003; 106:553 - 8; http://dx.doi.org/10.1002/ijc.11261; PMID: 12845651
- Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Brandsma J, et al. Molecular classification identifies a subset of human papillomavirus--associated oropharyngeal cancers with favorable prognosis. J Clin Oncol 2006; 24:736 - 47; http://dx.doi.org/10.1200/JCO.2004.00.3335; PMID: 16401683
- Licitra L, Perrone F, Bossi P, Suardi S, Mariani L, Artusi R, et al. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J Clin Oncol 2006; 24:5630 - 6; http://dx.doi.org/10.1200/JCO.2005.04.6136; PMID: 17179101
- Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst 2008; 100:261 - 9; http://dx.doi.org/10.1093/jnci/djn011; PMID: 18270337
- Gillison ML, D’Souza G, Westra W, Sugar E, Xiao W, Begum S, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst 2008; 100:407 - 20; http://dx.doi.org/10.1093/jnci/djn025; PMID: 18334711
- Psyrri A, Gouveris P, Vermorken JB. Human papillomavirus-related head and neck tumors: clinical and research implication. Curr Opin Oncol 2009; 21:201 - 5; http://dx.doi.org/10.1097/CCO.0b013e328329ab64; PMID: 19370803
- Balz V, Scheckenbach K, Götte K, Bockmühl U, Petersen I, Bier H. Is the p53 inactivation frequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2-11 and human papillomavirus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res 2003; 63:1188 - 91; PMID: 12649174
- Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med 2007; 357:2552 - 61; http://dx.doi.org/10.1056/NEJMoa073770; PMID: 18094376
- Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res 2008; 14:366 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-07-1402; PMID: 18223210
- Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993; 75:495 - 505; http://dx.doi.org/10.1016/0092-8674(93)90384-3; PMID: 8221889
- Münger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, et al. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 2001; 20:7888 - 98; http://dx.doi.org/10.1038/sj.onc.1204860; PMID: 11753671
- Alani RM, Münger K. Human papillomaviruses and associated malignancies. J Clin Oncol 1998; 16:330 - 7; PMID: 9440761
- Dowhanick JJ, McBride AA, Howley PM. Suppression of cellular proliferation by the papillomavirus E2 protein. J Virol 1995; 69:7791 - 9; PMID: 7494290
- Desaintes C, Demeret C, Goyat S, Yaniv M, Thierry F. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J 1997; 16:504 - 14; http://dx.doi.org/10.1093/emboj/16.3.504; PMID: 9034333
- Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci USA 2000; 97:12513 - 8; http://dx.doi.org/10.1073/pnas.97.23.12513; PMID: 11070078
- Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, Howley PM. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J 2000; 19:5762 - 71; http://dx.doi.org/10.1093/emboj/19.21.5762; PMID: 11060027
- Psyrri A, DeFilippis RA, Edwards AP, Yates KE, Manuelidis L, DiMaio D. Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res 2004; 64:3079 - 86; http://dx.doi.org/10.1158/0008-5472.CAN-03-3739; PMID: 15126344
- DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol 2003; 77:1551 - 63; http://dx.doi.org/10.1128/JVI.77.2.1551-1563.2003; PMID: 12502868
- Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, Hoppe-Seyler F. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 2003; 22:5938 - 45; http://dx.doi.org/10.1038/sj.onc.1206894; PMID: 12955072
- Hall AH, Alexander KA. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J Virol 2003; 77:6066 - 9; http://dx.doi.org/10.1128/JVI.77.10.6066-6069.2003; PMID: 12719599
- Sima N, Wang W, Kong D, Deng D, Xu Q, Zhou J, et al. RNA interference against HPV16 E7 oncogene leads to viral E6 and E7 suppression in cervical cancer cells and apoptosis via upregulation of Rb and p53. Apoptosis 2008; 13:273 - 81; http://dx.doi.org/10.1007/s10495-007-0163-8; PMID: 18060502
- Horner SM, DeFilippis RA, Manuelidis L, DiMaio D. Repression of the human papillomavirus E6 gene initiates p53-dependent, telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells. J Virol 2004; 78:4063 - 73; http://dx.doi.org/10.1128/JVI.78.8.4063-4073.2004; PMID: 15047823
- Ferris RL, Martinez I, Sirianni N, Wang J, López-Albaitero A, Gollin SM, et al. Human papillomavirus-16 associated squamous cell carcinoma of the head and neck (SCCHN): a natural disease model provides insights into viral carcinogenesis. Eur J Cancer 2005; 41:807 - 15; http://dx.doi.org/10.1016/j.ejca.2004.11.023; PMID: 15763658
- Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J Natl Cancer Inst 2009; 101:412 - 23; http://dx.doi.org/10.1093/jnci/djp017; PMID: 19276448
- Sterlinko Grm H, Weber M, Elston R, McIntosh P, Griffin H, Banks L, et al. Inhibition of E6-induced degradation of its cellular substrates by novel blocking peptides. J Mol Biol 2004; 335:971 - 85; http://dx.doi.org/10.1016/j.jmb.2003.10.079; PMID: 14698292
- Dymalla S, Scheffner M, Weber E, Sehr P, Lohrey C, Hoppe-Seyler F, et al. A novel peptide motif binding to and blocking the intracellular activity of the human papillomavirus E6 oncoprotein. J Mol Med (Berl) 2009; 87:321 - 31; http://dx.doi.org/10.1007/s00109-008-0432-1; PMID: 19099279
- D’Abramo CM, Archambault J. Small-molecule inhibitors of human papillomavirus protein - protein interactions. Open Virol J 2011; 5:80 - 95; http://dx.doi.org/10.2174/1874357901105010080; PMID: 21769307
- Yuan CH, Filippova M, Tungteakkhun SS, Duerksen-Hughes PJ, Krstenansky JL. Small-molecule inhibitors of the HPV16-E6 interaction with caspase 8. Bioorg Med Chem Lett 2012; 22:2125 - 9; http://dx.doi.org/10.1016/j.bmcl.2011.12.145; PMID: 22300659
- Dueñas-Gonzalez A, Cetina L, Coronel J, Cervantes-Madrid D. Emerging drugs for cervical cancer. Expert Opin Emerg Drugs 2012; 17:203 - 18; http://dx.doi.org/10.1517/14728214.2012.683409; PMID: 22530838
- Stern PL, van der Burg SH, Hampson IN, Broker TR, Fiander A, Lacey CJ, et al. Therapy of human papillomavirus-related disease. Vaccine 2012; 30:Suppl 5 F71 - 82; http://dx.doi.org/10.1016/j.vaccine.2012.05.091; PMID: 23199967
- Lin CJ, Grandis JR, Carey TE, Gollin SM, Whiteside TL, Koch WM, et al. Head and neck squamous cell carcinoma cell lines: established models and rationale for selection. Head Neck 2007; 29:163 - 88; http://dx.doi.org/10.1002/hed.20478; PMID: 17312569
- Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, et al. Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene 2005; 24:344 - 54; http://dx.doi.org/10.1038/sj.onc.1208225; PMID: 15531918
- Huang C, Hu X, Wang L, Lü S, Cheng H, Song X, et al. Bortezomib suppresses the growth of leukemia cells with Notch1 overexpression in vivo and in vitro. Cancer Chemother Pharmacol 2012; 70:801 - 9; http://dx.doi.org/10.1007/s00280-012-1953-4; PMID: 22996635
- Hutter G, Rieken M, Pastore A, Weigert O, Zimmermann Y, Weinkauf M, et al. The proteasome inhibitor bortezomib targets cell cycle and apoptosis and acts synergistically in a sequence-dependent way with chemotherapeutic agents in mantle cell lymphoma. Ann Hematol 2012; 91:847 - 56; http://dx.doi.org/10.1007/s00277-011-1377-y; PMID: 22231280
- Jabbar S, Strati K, Shin MK, Pitot HC, Lambert PF. Human papillomavirus type 16 E6 and E7 oncoproteins act synergistically to cause head and neck cancer in mice. Virology 2010; 407:60 - 7; http://dx.doi.org/10.1016/j.virol.2010.08.003; PMID: 20797753
- Strati K, Lambert PF. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res 2007; 67:11585 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-07-3007; PMID: 18089787
- Fujii T, Saito M, Iwasaki E, Ochiya T, Takei Y, Hayashi S, et al. Intratumor injection of small interfering RNA-targeting human papillomavirus 18 E6 and E7 successfully inhibits the growth of cervical cancer. Int J Oncol 2006; 29:541 - 8; PMID: 16865269
- Gu W, Putral L, Hengst K, Minto K, Saunders NA, Leggatt G, et al. Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral-vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes. Cancer Gene Ther 2006; 13:1023 - 32; http://dx.doi.org/10.1038/sj.cgt.7700971; PMID: 16810314
- Bross PF, Kane R, Farrell AT, Abraham S, Benson K, Brower ME, et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res 2004; 10:3954 - 64; http://dx.doi.org/10.1158/1078-0432.CCR-03-0781; PMID: 15217925
- Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003; 348:2609 - 17; http://dx.doi.org/10.1056/NEJMoa030288; PMID: 12826635
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al, Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352:2487 - 98; http://dx.doi.org/10.1056/NEJMoa043445; PMID: 15958804
- Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006; 24:4867 - 74; http://dx.doi.org/10.1200/JCO.2006.07.9665; PMID: 17001068
- Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res 2007; 67:4605 - 19; http://dx.doi.org/10.1158/0008-5472.CAN-06-3619; PMID: 17510386
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995; 80:293 - 9; http://dx.doi.org/10.1016/0092-8674(95)90412-3; PMID: 7834749
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7:673 - 82; http://dx.doi.org/10.1016/S1097-2765(01)00213-1; PMID: 11463391
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683 - 94; http://dx.doi.org/10.1016/S1097-2765(01)00214-3; PMID: 11463392
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288:1053 - 8; http://dx.doi.org/10.1126/science.288.5468.1053; PMID: 10807576
- Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 2002; 4:842 - 9; http://dx.doi.org/10.1038/ncb866; PMID: 12402042
- Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 2003; 4:371 - 81; http://dx.doi.org/10.1016/S1535-6108(03)00272-1; PMID: 14667504
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010 - 4; http://dx.doi.org/10.1126/science.1092734; PMID: 14963330
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6:443 - 50; http://dx.doi.org/10.1038/ncb1123; PMID: 15077116
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577 - 90; http://dx.doi.org/10.1016/S1097-2765(03)00050-9; PMID: 12667443
- Zang Y, Thomas SM, Chan ET, Kirk CJ, Freilino ML, DeLancey HM, et al. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin Cancer Res 2012; 18:5639 - 49; http://dx.doi.org/10.1158/1078-0432.CCR-12-1213; PMID: 22929803