Abstract
The synthesis and degradation of hBora is important for the regulation of mitotic entry and exist. In G2 phase, hBora can complex with Aurora A to activate Plk1 and control mitotic entry. However, whether the post-translational modification of hBora is relevant to the mitotic entry still unclear. Here, we used the LC-MS/MS phosphopeptide mapping assay to identify 13 in vivo hBora phosphorylation sites and characterized that GSK3β can interact with hBora and phosphorylate hBora at Ser274 and Ser278. Pharmacological inhibitors of GSK3β reduced the retarded migrating band of hBora in cells and diminished the phosphorylation of hBora by in vitro kinase assay. Moreover, as well as in GSK3β activity-inhibited cells, specific knockdown of GSK3β by shRNA and S274A/S278 hBora mutant-expressing cells also exhibited the reduced Plk1 activation and a delay in mitotic entry. It suggests that GSK3β activity is required for hBora-mediated mitotic entry through Ser274 and Ser278 phosphorylation.
Keywords: :
Introduction
The mitotic entry is triggered by the activation of Cyclin-B1/Cdk1 complex.Citation1 In late G2, two independent studies demonstrate that Aurora A, in complex with its cofactor hBora, phosphorylates Plk1 at Thr210 and promotes mitotic entry either in unperturbed growthCitation2 or after recovery from a DNA-damage checkpoint.Citation3 Plk1 can activate the Cdk1-activating Cdc25 phosphataseCitation4 and induce the ubiquitin-dependent degradation of the Cdk1-repressor Wee1 by β-TrCP.Citation5 Altogether, it suggests that Plk1 exerts its effects on the regulation of G2/M transition to promote mitotic entry, and pT210-Plk1 status can be used as an G2/M indicator.
A number of recent works highlight the role of hBora in cell cycle progression of mitotic entry and exit.Citation2,Citation6,Citation7 hBora expression is low in G1/S boundary but increased in late S phase, accumulates in the G2 phase and then hBora is phosphorylated at Ser252, which is a Cdk1-primed PBD-docking site of Plk1 and degraded through β-TrCP-dependent proteasomal degradation in mitosis.Citation7 hBora exhibited a retarded migrating motility during mitosis, which can be collapsed by alkaline phosphatase treatment, reflecting the highly phosphorylated states of hBora.Citation7 However, whether the post-translational modification of hBora is involved in controlling mitotic entry through phosphorylation remains unclear.
Glycogen synthase kinase 3 (GSK3) is a multi-functional serine/threonine kinase that performs a role in several signaling pathways involved in the regulation of cell fate. GSK3 has been extensively studied in several physiological processes, such as cell proliferation, glycogen metabolism and neuron degeneration.Citation8 For cell cycle regulation as well as to determine the turnover of cyclin-D1 to regulate G1-S progression,Citation9 GSK3 also involves in interphase microtubule dynamics and chromosome alignment.Citation10,Citation11 Interestingly, previous studies show that GSK3 inhibitors can delay mitotic entry and exit in Hela cellCitation12 and G2-M accumulation in glioblastoma cells.Citation13-Citation15 Moreover, another independent study shows that inhibit GSK3 activity is associated with G2/M cell cycle arrest and an increased expression of phospho-Cdk1(Tyr15) but does not have an apparent effect on G1/S progression,Citation15 suggesting that GSK3 activity is relevant to Cyclin-B1/Cdk1-mediated mitotic entry.
Whereas the mechanism of GSK3 in mitotic entry remains unclear, the results of bioinformatic prediction indicate that hBora contains 23 sites with the consensus T/SXXXS/T phosphoacceptor sequence for GSK3β.Citation16 We therefore speculate whether or not GSK3β activity is associated with hBora-mediated mitotic entry. In this study, we have explored the functional regulation of hBora and GSK3β interaction in mitotic entry. We identify 13 novel hBora in vivo phosphorylation sites by LC-MS/MS and characterize that Ser274 and Ser278 are GSK3β-mediated phopshorylation sites. hBora can interact with GSK3β in vitro and in vivo. S274A/S278A hBora mutant exhibits reduced slowly migrating bands, which is similar to wild-type hBora in GSK3β activity-inhibited cells. Furthermore, using shRNA to silence endogenous GSK3β expression displayed the delayed activation of Plk1 and mitotic entry, that is similar to S274A/S278A hBora mutant-expressing and GSK3β activity-inhibited cells. Taken these together, our findings indicate that GSK3β has a role in mitotic entry through hBora.
Results
Identification of in vivo hBora phosphorylation sites
hBora can cooperate with Aurora A to activate Plk1 at the G2/M transition. Therefore, we analyzed the phosphorylation status of hBora in G2/M phase. The results of showed that hBora exhibited a retarded migrating motility in nocodazole-treated G2/M cells, which can be collapsed by calf intestinal alkaline phosphatase (CIP) treatment. Moreover, together with the fact that immunoreactivity of pSer/pThr-Pro was markedly reduced in CIP-treated Flag-hBora experiments, this reflected that hBora is highly phosphorylated in G2/M phase. Next, to identify novel hBora phosphorylation sites in vivo, LC-MS/MS-based phosphopeptide mapping analysis was used. A total of 13 phosphorylation sites were identified from hBora in G2/M-arrested HEK293T cells (Table S1). One of the identified hBora phosphorylation sites is Ser252, which has been characterized as a potential phosphorylation site by Cdk1 kinase activity in cells,Citation7 suggesting that the results of phosphopeptide mapping analysis are reliable and usable. Next, we examined the biological significance of these identified phosphorylation sites. By site-directed mutagenesis and western blotting assay, we found that S274A/S278A hBora mutant showed reduced slowly migrating bands rather than other hBora mutants (). This suggests that S274A/S278A hBora mutant reduced the highly phosphorylated status of hBora in G2/M phase, and Ser274 and Ser278 phosphorylation may act as an important initiated phosphorylation sites. showed the LC-MS/MS spectra of phosphorylated peptides on Ser274 and Ser278 of hBora.
Figure 1. Ser274 and Ser278 are critical in vivo hBora phosphorylation sites. (A) hBora is highly phosphorylated in G2/M phase. Nocodazole-synchronized Flag-hBora-expressing HEK293T cell extracts were incubated with or without calf intestinal phosphatase (CIP) and then subjected to IP-western assay using anti-Flag antibody and probed with anti-pSer/pThr-Pro antibody. (B) S274A/S278A hBora mutant reduces the retarded migrating bands. The indicated hBora mutants were expressed in HEK293T cells and then synchronized by nocodazole. The cell extracts were analyzed by western blotting. (C) MS/MS spectrum of the phosphopeptide 274-SPTFSP-279 from hBora in G2/M-arrested HEK293T cells. Ser274 and Ser278 of hBora were identified as in vivo phosphorylation sites by LC-MS/MS-based phosphopeptide mapping assay. (D) A GSK3β inhibitor markedly reduces the retarded migrating bands. Flag-hBora-expressing HEK293T cells were treated with the indicated kinase inhibitors and subjected to western blotting. (E) A schematic representation of possible GSK3β phosphorylation sites on hBora by three independent phosphorylation site prediction databases (GPS2.1, PPSP and NetPhosK1.0). The sequence S274PTFS278P of hBora is similar to the putative GSK3β conserved phosphorylation sequences (Ser/Thr-X-X-X-Ser/Thr).
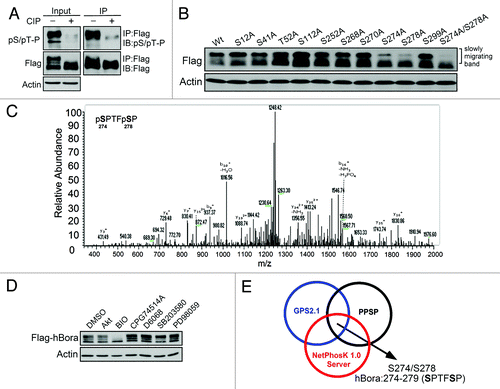
GSK3β is selected as a candidate kinase for Ser274 and Ser278 phosphorylation of hBora
Subsequently, we attempted to indentify the plausible kinase(s) that can phosphorylate Ser274 and Ser278 in G2/M phase, a phosphorylation bioinformatic prediction approach was used. The results of bioinformatic prediction revealed that GSK3β, Akt, Cdk1, Aurora A, p38 and MEK1 are potential kinases to phosphorylate hBora in cells (Table S1). GSK3β was then selected for the following reasons. First, by using the indicated kinase inhibitors, Akt inhibitor IV (Akt), BIO (GSK3β), CGP74514A (Cdk1), D6068 (Aurora A), SB203580 (p38) and PD98059 (MEK1) in hBora-expressing HEK293T cells, none of these pharmacological inhibitors but GSK3β inhibitor BIO showed significant ability to reduce the slowly migrating retarded bands (). Second, the sequence of hBora Ser274 to Ser278, S274PTFS278P, is highly similar to putative GSK3β conserved phosphorylation sequences (Ser/Thr-X-X-X-Ser/Thr). Besides, Ser274 and Ser278 were also identified as potential GSK3β phosphorylation sites by three independent bioinformatic analyses with high scores (). Third, GSK3β activity-inhibited cells exhibited a delay in mitotic entry that is similar to hBora-depleted cells, suggesting hBora and GSK3β play the same role in regulation of mitotic entry. Taken together, these evidences hypothesize that GSK3β activity is relevant to Ser274 and Ser278 phosphorylation of hBora and that can regulate mitotic entry.
GSK3β interacts with hBora in vitro and in vivo
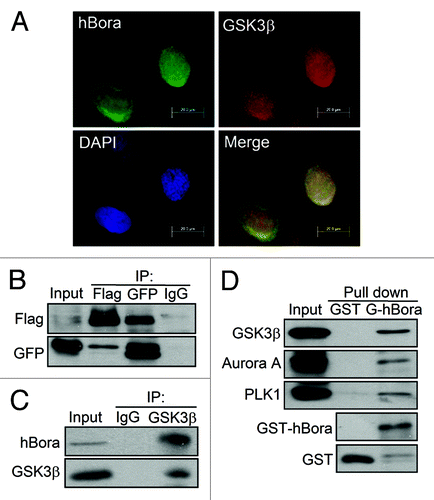
Accordingly, biological characterization of the hBora-GSK3β interaction and phosphorylation events, including immunofluorescence microscopy analysis, immunoprecipitation assay, GST pull-down assay and in vitro GSK3β kinase assay were performed to address whether GSK3β phosphorylates hBora and control hBora-mediated mitotic entry. The immunofluorescence assay was performed to verify the co-localization of GSK3β and hBora in G2/M cells (). Moreover, this interaction was confirmed in a transient transfection experiment by coimmunoprecipitation of GFP-GSK3β with Flag-hBora reciprocally but not with IgG control () or readily confirmed by endogenous hBora co-precipitated with the anti-GSK3β antibody beads. This interaction is specific, as control IgG precipitated neither GSK3β nor hBora (). To further corroborate the interaction between hBora and GSK3β, it was confirmed by GST pull-down assay (). Purified GST-hBora was incubated with nocodazole-arrested U2OS cell lysates and analyzed by western blotting. The results showed that endogenous GSK3β binds to GST-hBora protein in vitro. Both Aurora A and Plk1 have been proved as hBora binding proteins,Citation2,Citation7 Aurora A and Plk1 were also detected in the pull down as positive controls. Collectively, there is agreement between immunofluorescence microscopy analysis; in vivo and in vitro binding results validated that GSK3β and hBora interact with each other in cells.
Figure 2. GSK3β interacts with hBora in vivo and in vitro. (A) The colocalization of GSK3β and hBora in G2/M U2OS cells. At 14 h release from thymidine, U2OS cells were fixed and subjected to immunofluorescence staining with GSK3β (red), hBora (green) and DAPI (nucleus, blue), followed by fluorescence microscopy analysis. (B) GSK3β interacts with hBora in vivo. Flag-hBora and GFP-GSK3β-coexpressing HEK293T cell extracts were subjected to immunoprecipitation assay using anti-Flag or anti-GFP antibodies. IgG was used as a negative control. (C) Endogenous hBora interacts with GSK3β. As described in (B), Nocodazole-synchronized U2OS were subjected to immunoprecipitation assay using anti-GSK3β antibody. (D) hBora interacts with GSK3β in vitro. HEK293T cell extracts were subjected to GST pull-down with either GST or GST-hBora. The bound proteins were detected by western blotting using the indicated antibodies.
GSK3β phosphorylates Ser274 and Ser278 of hBora
To explore whether hBora is a potential substrate of GSK3β by in vitro kinase assay, purified His-GSK3β activity was assayed using His-tau protein as positive substrate (, left panel). GST-hBora recombinant proteins (right panel) were incubated in kinase reaction buffer containing [γ-32P] ATP with His-GSK3β and in the absence or presence of GSK3β inhibitor-BIO, respectively. Analysis of the results by SDS-PAGE and phosphate incorporation was visualized by autoradiography. Both of tau and hBora could be phosphorylated by GSK3β, whereas the radioactivity of the same protein pre-incubated with BIO was barely detectable (). Moreover, the results of bioinformatic prediction showed that GSK3β is suggested to phosphorylate hBora at Ser274 and Ser278 residues of S274PTFS278P region, which are immediately preceded by a proline residue. It is therefore to speculate that GSK3β-mediated hBora phosphorylation can be recognized by a pSer/pThr-Pro antibody. To address the question, in vitro hBora phosphorylation by GSK3β kinase were subjected to western blotting using a pSer/pThr-Pro antibody. showed that hBora pSer/pThr-Pro signal was detected and with a time-dependent increase at the indicated times after incubated with GSK3β kinase, whereas the signal was barely detected in BIO pre-treated condition.
Figure 3. GSK3β phosphorylates hBora at Ser274 and Ser278. (A) hBora is a potential substrate of GSK3β. GST-hBora protein was subjected to in vitro GSK3β kinase assay, which was incubated in the absence or presence of His-GSK3β or BIO-preincubated His-GSK3β, respectively. The samples were separated by SDS-PAGE and then followed by autoradiography. His-tau was used as a positive control to represent His-GSK3β kinase activity. (B) GSK3β phosphorylates hBora at Ser/Thr-Pro motif. GST-hBora protein was subjected to GSK3β kinase assay at the indicated time and then analyzed by western blotting using pSer/pThr-Pro and hBora antibodies. (Cand D) Ser274 and Ser278 of hBora are critical GSK3β phosphorylation sites. GST-hBora and its mutant proteins were subjected to GSK3β kinase assay, as described in (A) or analyzed by western blotting as (B).
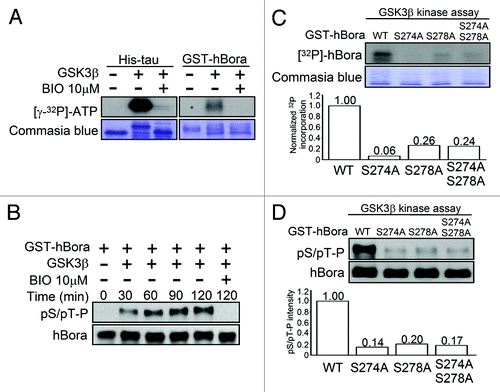
As stated above, the results enhanced the possibility of GSK3β phosphorylating hBora at Ser274 and Ser278 immediately preceding a proline residue. To corroborate the possibility, mutant recombinant hBora protein by mutating Ser274 and Ser278 to Ala, either singly or doubly, were subjected to in vitro GSK3β kinase assay. Compared with WT-hBora, mutations of Ser274, Ser278 or both markedly blocked hBora phosphorylation by GSK3β with 99%, 74% and 76% reduction, respectively (), suggesting that Ser274 and Ser278 are critical GSK3β phosphorylation sites. In addition, S274A, S278A or both of hBora mutations failed to stain with pSer/pThr-Pro antibody after GSK3β kinase assay (), the reduction amount of pSer/pThr-Pro intensity were 86%, 80% and 83%, respectively. Collectively, we conclude that GSK3β can interact with and phosphorylate hBora at Ser274 and Ser278 in vitro and in vivo.
GSK3β-mediated hBora phosphorylation is required for mitotic entry
The next question is to investigate the physiological significance of the GSK3β-hBora interaction. S274A/S278A mutant hBora combined with manipulation of GSK3β activity using pharmacological inhibitor and genetic inhibition of GSK3β by kinase dead GSK3β and shRNA to silence GSK3β expression were used to explore the GSK3β-mediated hBora phosphorylation in regulating mitotic entry. First, HEK293T cells were transfected with Flag-hBora and then used BIO to inhibit GSK3β activity; the results showed that inhibition of GSK3β activity resulted in a striking reduction of upshifted form hBora (, right panel). It suggested that the decreased upshifted bands were caused by the reduction of Ser274 and Ser278 phosphorylation in GSK3β activity-impaired cells. Similarly, S274A/S278A hBora mutant also exhibited a reduction of upshifted bands (, left panel, upper) and compared with WT-hBora, pSer/pThr-Pro immunoreactivity failed to stain the upshifted form of hBora by IP-western analysis (left panel, lower). Collectively, together with the results of GSK3β interacts with and phosphoryaltes hBora ( and ), it suggests GSK3β-dependent S274 and S278 phosphorylation of hBora contributed the upshifted migration in vivo.
Figure 4. GSK3β-mediated hBora phosphorylation is required for mitotic entry. (A) The slowly migrating retarded bands of hBora are reduced in the S274A/S278A mutant and in BIO-treated cells. Flag-hBora-expressing HEK293T cells were treated with BIO or DMSO as a vehicle control (right panel). Flag-hBora or Flag-S274A/S278A mutant hBora-expressing HEK293T cells were subjected to an IP-western assay (left panel). All samples were subjected to western blotting using the indicated antibodies. (B) S274A/S278A mutant hBora delays the activation of Plk1. p-Thr210 status of Plk1 were examined in Flag-hBora or Flag-S274A/S278A hBora-expressing HEK293T cells at the indicated times after thymidine release (Thy release) by western blotting and plotted after normalizing the indicated samples to Plk1 and actin (p-T210 Plk1 of hBora mutant at 8 h time point is set at 1). (C) Thymidine-released cell extracts (16 h) of control and SB415286-treated U2OS (10 μM) were collected and subjected to western blotting using MPM2 to validate the mitotic index and p-Thr210 Plk1 to represent G2/M transition (left panel). In right panel, control and GSK3β activity-inhibited U2OS cells were used to examine the ability of mitotic entry. Phase images of cells were acquired at 16 h post thymidine release. Note the difference in the percentage of mitotic round cells between control and GSK3β activity-inhibited samples. The amount of mitotic cells markedly reduced in GSK3β activity-inhibited cells. (D) The inhibition of GSK3β activity delays mitotic entry. Thymidine-released U2OS cells were treated with BIO (0, 1 or 2 μM) at 6 h into this release, were trapped in mitosis by nocodazole for another 10 h and then analyzed by western blotting. p-Thr210 Plk1 (G2/M indicator) and mitotic index is calculated by quantifying the relative intensity of MPM2 by using actin for normalization. U2OS cells showed a dose-dependent reduction of phospho-Thr210 Plk1 and MPM2 intensity. The results are from three individual experiments, and the bars indicate the SD *p < 0.05. (E) Loss of GSK3β expression delays the activation of Aurora A and Plk1 that results in a delay in mitotic entry. GSK3β knockdown and scrambled control cells were released form thymidine for 12, 14, 16 h. The cell extracts were analyzed by western blotting using the indicated antibodies. p-T288 Aurora A and p-Thr210 Plk1 act as G2/M indicator and the intensity of MPM2 represents the mitotic index. The mitotic index is calculated by quantifying the relative intensity of MPM2 by using actin for normalization at the thymidine-released time points (12, 14, 16 h).
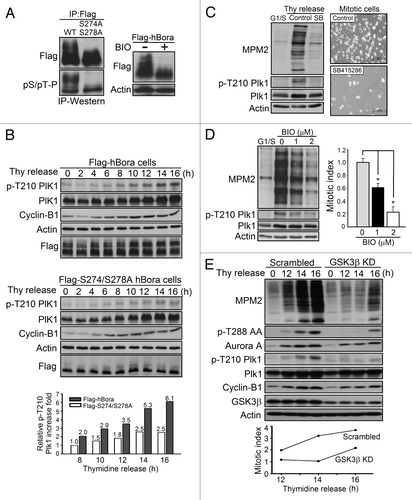
hBora is demonstrated as a G2/M regulator to control mitotic entry,Citation2,Citation3 and the mechanisms by which inhibition of GSK3 activity delays mitotic entryCitation12,Citation13,Citation15 through hBora remains unclear. Taken together, we next to investigate that whether GSK3β-mediated hBora phosphorylation is required for Plk1 activation and control mitotic entry. To address the question, Plk1-Thr210 phosphorylation profiles of Flag-hBora and Flag-S274A/S278A hBora mutant-expressed HEK293T cell extracts were examined at the indicated times following release from thymidine block. The results in showed that WT-hBora formed a retarded electrophoretic mobility shift, whereas S274A/S278A mutant hBora expressing cells reduced the upshifted forms of hBora at any time following release from thymidine block. In addition, phospho-Thr210 of Plk1 (G2/M indicator) was markedly reduced in S274A/S278A mutant-expressing cells when compared with WT-hBora-expressing cells. At 16 h of thymidine release, Plk1-Thr210 phosphorylation levels showed a 6.1-fold increase in WT-hBora, whereas in S274A/S278A mutant cells only had 2.5-fold increase, suggesting that S274A/S278A hBora mutant fails to activate Plk1 (, bottom panel).
Because we reasoned that GSK3β is responsible for hBora S274/S278 phosphorylation to control mitotic entry, silencing endogenous GSK3β expression by shRNA, overexpressing a dominant-negative mutant, GSK3β-R96A, or using pharmacological inhibitors to inhibit GSK3β activity in U2OS cells would delay mitotic entry. MPM2 antibody recognizes phosphoproteins in mitotic cells, which can be used an index for mitotic entry. The results showed that all GSK3β inhibitor-treated cells, SB415286, BIO and lithium chloride significantly reduced the MPM2 signal. In , after 16 h release from a thymidine blockade, the percentage of mitotic cells was markedly reduced in the GSK3β activity-inhibited cells when compared with the control cells (right panel), and phospho-Thr210 Plk1 levels were also reduced in GSK3β activity-inhibited cells (left panel). Moreover, U2OS cells treated with 1 or 2 μM BIO showed a dose-dependent reduction of MPM2 index in 40% and 80% and in Plk1 activation, respectively (). The similar results of inhibited GSK3β activity that can delay mitotic entry were also observed in lithium chloride-treated cells (Fig. S1A). Furthermore, to further confirm that these results were not caused by the off-target effects of pharmacological inhibitors, overexpression of a dominant-negative mutant, GSK3β-R96A, and knockdown GSK3β expression by shRNA were used. As shown in , scrambled control and GSK3β-knockdown cells were synchronized by thymidine and released for the indicated times (12, 14, 16 h). Compared with scrambled control cells, GSK3β knockdown cells exhibited a marked reduction of mitotic index (MPM2 intensity) at the same thymidine release time point, as well as showed in GSK3β-R96A-expressing cells, which exhibited 48% reduction of MPM2 after 16 h thymidine release (Fig. S1B). In addition, GSK3β-knockdown cells also expressed a marked delay in the activation of Aurora A (phospho-T288 AA) and Plk1 (phospho-T210 Plk1), suggesting that loss of GSK3β expression delays Aurora A and Plk1 activation that results in a delay in mitotic entry. Collectively, together with S274A/S278A, hBora mutant also exhibited a reduced MPM2 staining (Fig. S1C), and GSK3β interacts with hBora, which indicates that hBora S274/S278 phosphorylation can activate Plk1 and promote mitotic entry that is regulated by GSK3β activity.
Discussion
This study focuses on elucidating the relationship between GSK3β and hBora during G2/M transition. We exhibited a chemical biology approach using small-molecule kinase inhibitors to inactivate GSK3β and then analyzed the effect of its inactivation on entry into mitosis. We also followed the kinase activity directly through analysis of hBora phosphorylation using in vitro GSK3β kinase assay. We found the in vivo phosphoryaltion sites of hBora at Ser274 and Ser278 by LC-MS/MS-based phosphopeptides mapping analysis, which are characterized as GSK3β phosphorylation sites, and that is regulated by GSK3β-hBora interaction. Moreover, as well as in S274A/S278A hBora mutant, the inhibition of GSK3β with a small-molecule kinase and specific silencing of GSK3β expression by shRNA can prevent the activation of Plk1 and delay the mitotic entry. Together, our data show that GSK3β plays a role in Plk1-mediated mitotic entry through regulating the hBora/Aurora A complex.
GSK3 is heavily involved in many important signaling cascades that may regulate glucose homeostasis and the development of insulin resistance;Citation17 very little is known about the effect of GSK3 on cell cycle regulation. GSK3β activity and distribution is showed to relate to cell cycle progression. In G1 phase, GSK3β is predominantly located in cytoplasm and then enters the nucleus during the S phase to phosphorylate cyclin-D1, and that results in the export of cyclin-D1 from the nucleus for degradation to control S phase progression.Citation9 Our results showed that GSK3β can colocalize with a G2/M regulator-hBora (), suggesting a novel function of GSK3β in G2/M regulation. Several independent studiesCitation12,Citation13,Citation15 showed that inhibition of GSK3 activity by lithium chloride and SB415286 decreased cell proliferation, does not apparently effect on G1/S progression, but is associated with a elevated expression of phospho-Cdk1 at Tyr15 and G2/M accumulation or G2/M arrest of cell cycle instead. It suggests that GSK3 activity is involved in regulating the upstream of Cyclin-B1/Cdk1 complex activity and GSK3 exerts its effect on mitotic entry. Indeed, our findings reveal, in part, the mechanism for GSK3β-mediated mitotic entry through the regulation of hBora. GSK3β interacts with and phosphorylates hBora at Ser274 and Ser278, which is involved in Thr210 phosphorylation and activation of Plk1.
Previous studies and ours showed the expression of Flag-hBora in HEK293T cells resulted in the appearance of an upshifted band in G2/M cells, and this upshifted band was sensitive to phosphatase treatment and absent in the S274A/S278A hBora mutant expressing cells. Interestingly, the upshifted band of hBora does not absent in other mutants, such as S252A. It reflects that the upshifted band is a phosphorylated form of hBora and Ser274/Ser278 residues may act as critical phosphorylation initiation sites. Notably, together with Ser274/Ser278 being characterized as GSK3β phosphorylation sites, it is reasonable to speculate that after GSK3β-mediated phosphorylation of hBora, hBora may be further phosphorylated by other mitotic kinases, such as Ser252 by Cdk1 and Ser497 and Thr501 by Plk1. However, the detailed mechanisms remains to be further examined.
In addition to metabolic diseases, GSK3 is also linked with Alzheimer disease.Citation18 Memory impairment, β-amyloid plaque formation, tau hyperphosphorylation and apoptotic neuronal loss have been addressed as the main histopathological hallmarks of this disease. Increasing evidence shows that ectopic cell cycle re-entry can also be considered a neuropathological marker for AD.Citation19 Previous study showed that a mitotic phosphoprotein monoclonal-2 (MPM-2) antibody, which strongly reacts with mitotic protein extracts in AD patients’ brains, is concomitant with aberrant expression of Cyclin-B1/Cdk1 in degenerating neurons of Alzheimer disease brain but absent in the healthy brain.Citation20 However, the mechanism of cell cycle re-entry remains unclear. Here, we reported that GSK3β activity is relevant to mitotic entry through interacting and regulating hBora, and that results in Cyclin-B1/Cdk1 activation. Together with the overactivation of GSK3 in Alzheimer disease, it is speculated that hBora is one target of GSK3-mediated processes in Alzheimer disease to promote cell cycle re-entry and increase mitotic events.
Materials and Methods
Cells culture, transfection, synchronization
Human osteosarcoma U2OS and HEK293T cells were all grown at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium, supplemented with penicillin-streptomycin (100 IU/ml and 100 mg/ml, respectively) and 10% fetal bovine serum. Transfection was performed by electroporation (Neon® Transfection System) for U2OS cells and by using Lipofectamine 2000 (Invitrogen) for HEK293T cells, according to the manufacturer’s instructions. The following plasmids were used for transfections: human hBora cDNA plasmid-BC056876 was purchased from Open Biosystems and subcloned into pCMV-Flag and pEGFP-vectors. pCMV-hBora-S41A, hBora-S112A, hBora-S252A, hBora-S268A, hBora-S270A, hBora-S274A/S278A, hBora-S270A/S274A/S278A, hBora-S299A, peGFP-S274A/S278A were constructed by site-directed mutagenesis. To generate the scramble control and stable GSK3β-knockdown cell lines, U2OS cells were transfected with pGIPZ non-silencing shRNA control vector and GSK3β shRNA plasmids that were obtained from Open Biosystems. After 48 h, transfected cells were treated with 2 ng/ml puromycin for 3 weeks to select stable GSK3β knockdown cells.
For synchronization, cells were treated with 2.5 mM thymidine for 24 h to block cells at the G1/S boundary, and then cells were subsequently washed three times with HBSS and released into fresh medium, after which samples were taken at regular intervals for western blotting analysis. Alternatively, to collect cells in G2/M phase, cells were synchronized by 200 ng/ml nocodazole for 14 h. For the examination of GSK3β activity in mitotic entry, U2OS cells were synchronized at the G1/S boundary and released from the thymidine. To observe the ability of mitotic entry, at 6 h release, cells were treated with GSK3β inhibitor and nocodazole to trap mitotic cells.
LC-MS/MS phosphopeptide mapping analysis
HEK293T cells were transfected with Flag-hBora and then synchronized in G2/M phase by 200 ng/ml Nocodazole for 14 h. Flag-hBora was purified using anti-Flag antibody and the immunoprecipitates were separated by SDS-PAGE. Gel bands were excised, and tryptic digestion was performed and for identification of phosphorylation sites within these tryptic digests,Citation21 phosphorylated peptides were first identified by MALDI-TOF mass spectrometry.Citation22 The exact localization of the phosphorylated residues within the peptides was determined by MS/MS based sequencing using a quadrupole time-of-flight (QTOF) mass spectrometer to identify the in vivo phosphorylation sites of hBora.
Production of recombinant proteins, in vitro kinase assay, antibodies and reagents
GST-fusion proteins (WT-hBora and its mutants) and His-tagged GSK3β were produced in BL21 bacteria and affinity purified on Glutathione agarose beads following the manufacturer’s instructions. In vitro kinase assays for recombinant proteins were performed in the GSK3β kinase buffer: 80 mM Pipes, pH 6.8/0.5 mM, MgCl2/1mM EGTA, 5 mM β-glycerophosphate and 1 mM DTT. Kinase reactions were performed at RT at the indicated times (0, 30, 60, 90, 120 min) in buffer supplemented with 10 μM ATP and 2 μCi [γ-32P] ATP. Reactions were stopped by the addition of 2× sample buffer and heating to 100°C. Reaction products were visualized by SDS-PAGE followed by autoradiography or subjected to western blotting analysis using pSer/pThr-Pro specific-MPM2 antibody.
The following antibodies were obtained from commercial sources: anti-phospho-Ser10 Histone H3, (Cell Signaling Technology); anti-actin, anti-Flag (Sigma-Aldrich); anti-Cyclin-B1, anti-Plk1, anti-GFP (Santa Cruz Biotechnology, Inc.), anti-Aurora A, anti-hBora (Abcam); anti-GSK3β, anti-phospho-Thr210 Plk1 (BD), anti-phospho-Thr288 Aurora A (Cell signaling) and anti-MPM2 (Millipore). Akt inhibitor IV (B2311), BIO (B1686), D6068, CGP74514A (C3353), SB203580 (S8307), SB415286 (S3567), nocodazole (M1404) and thymidine (T1895) were purchased from Sigma-Aldrich, PD98059 (513000) were purchased from Merck Millipore for the experimental usage.
Protein binding assays
GST pull-down, immunoprecipitation analyses were performed as previously described.Citation23 Relevant proteins were expressed in HEK293T or U2OS cells by transient transfection, followed by lysis in RIPA buffer containing protease and phosphatase inhibitor. Lysates were centrifuged prior to binding assays. The GST, GST-hBora recombinant proteins were bound on GSH-agarose beads and then incubated with cell lysates at 4°C for 2–3 h, and then beads were washed four times in 1 ml of binding buffer (50 mM HEPES, 50 mM TRIS-HCl, 100 mM NaCl, 0.5% Tween 20, 1 mM DTT, pH7.4). Subsequently, proteins were solubilized in 2× SDS sample buffer and separated by SDS-PAGE. Bound proteins were analyzed by western blotting.
For immunoprecipitations, 500 μg-1mg of total lysate protein was used per sample, and binding was performed using the indicated antibodies (2–4 μg), including anti-Flag, anti-GFP, anti-GSK3β and anti-hBora antibodies. Binding was performed by preincubating antibodies with Protein G or Protein A beads (Millipore), and then the beads were incubated with cellular supernatants for 2–3 h at 4°C. The precipitated proteins were washed three to five times in binding buffer (50 mM HEPES, 150 mM NaCl, 0.5% NP-40, pH 7.4) and subjected to western blotting.
Immunofluorescence microscopy analysis
Immunofluorescence experiments were performed as following description. U2OS cells were grown on coverslips. For formalin fixation, cells were fixed for 15 min at RT in a 3.7% formalin solution before permeablizing by PBS with 0.4% Triton-X-100 for 10 min at RT. Coverslips were then washed with PBS and blocked with 2% BSA in PBS. Incubation of both primary and secondary antibodies was done in PBS for overnight at 4°C. Primary antibodies used were anti-GSK3β, anti-hBora. Secondary fluorescence antibodies were anti-mouse Alexa-488 (green) and anti-rabbit Alexa-633 (red). To visualize DNA, the cells were stained with 4',6-diamidino-2-phenylindole (DAPI), and the cover glasses are mounted to microscope slides by mounting solution. Finally, the cover glass is fixed by nail varnish and observed by a fluorescent microscope or laser scanning confocal microscope.
Additional material
Download Zip (956.9 KB)Acknowledgments
This study was supported in part by National Science Council grant (NSC 101-2325-B-006-011), National Health Research Institutes grant of Taiwan (NHRI-EX101-10152SI) and a grant from Chi Mei Medical Center (CMNCKU9901).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
Yu-Cheng Lee designed the study, drafting and writing the manuscript. Po-Chi Liao provided LC/MS/MS-based phosphopepetides mapping analysis. Yih-Cherng Liou reviewed the manuscript. Michael Hsiao, Chi-Ying Huang and Pei-Jung Lu gave conceptual advice, provided materials and reviewed the manuscript.
References
- Nurse P. Universal control mechanism regulating onset of M-phase. Nature 1990; 344:503 - 8; http://dx.doi.org/10.1038/344503a0; PMID: 2138713
- Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320:1655 - 8; http://dx.doi.org/10.1126/science.1157425; PMID: 18566290
- Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455:119 - 23; http://dx.doi.org/10.1038/nature07185; PMID: 18615013
- van Vugt MA, Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005; 24:2844 - 59; http://dx.doi.org/10.1038/sj.onc.1208617; PMID: 15838519
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, et al. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci USA 2004; 101:4419 - 24; http://dx.doi.org/10.1073/pnas.0307700101; PMID: 15070733
- Seki A, Coppinger JA, Du H, Jang CY, Yates JR 3rd, Fang G. Plk1- and beta-TrCP-dependent degradation of Bora controls mitotic progression. J Cell Biol 2008; 181:65 - 78; http://dx.doi.org/10.1083/jcb.200712027; PMID: 18378770
- Chan EH, Santamaria A, Silljé HH, Nigg EA. Plk1 regulates mitotic Aurora A function through betaTrCP-dependent degradation of hBora. Chromosoma 2008; 117:457 - 69; http://dx.doi.org/10.1007/s00412-008-0165-5; PMID: 18521620
- Forde JE, Dale TC. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci 2007; 64:1930 - 44; http://dx.doi.org/10.1007/s00018-007-7045-7; PMID: 17530463
- Takahashi-Yanaga F, Sasaguri T. GSK-3beta regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal 2008; 20:581 - 9; http://dx.doi.org/10.1016/j.cellsig.2007.10.018; PMID: 18023328
- Wakefield JG, Stephens DJ, Tavaré JM. A role for glycogen synthase kinase-3 in mitotic spindle dynamics and chromosome alignment. J Cell Sci 2003; 116:637 - 46; http://dx.doi.org/10.1242/jcs.00273; PMID: 12538764
- Ciani L, Krylova O, Smalley MJ, Dale TC, Salinas PC. A divergent canonical WNT-signaling pathway regulates microtubule dynamics: dishevelled signals locally to stabilize microtubules. J Cell Biol 2004; 164:243 - 53; http://dx.doi.org/10.1083/jcb.200309096; PMID: 14734535
- Tighe A, Ray-Sinha A, Staples OD, Taylor SS. GSK-3 inhibitors induce chromosome instability. BMC Cell Biol 2007; 8:34; http://dx.doi.org/10.1186/1471-2121-8-34; PMID: 17697341
- Korur S, Huber RM, Sivasankaran B, Petrich M, Morin P Jr., Hemmings BA, et al. GSK3beta regulates differentiation and growth arrest in glioblastoma. PLoS ONE 2009; 4:e7443; http://dx.doi.org/10.1371/journal.pone.0007443; PMID: 19823589
- Dickey A, Schleicher S, Leahy K, Hu R, Hallahan D, Thotala DK. GSK-3β inhibition promotes cell death, apoptosis, and in vivo tumor growth delay in neuroblastoma Neuro-2A cell line. J Neurooncol 2011; 104:145 - 53; http://dx.doi.org/10.1007/s11060-010-0491-3; PMID: 21161565
- Pizarro JG, Folch J, Esparza JL, Jordan J, Pallàs M, Camins A. A molecular study of pathways involved in the inhibition of cell proliferation in neuroblastoma B65 cells by the GSK-3 inhibitors lithium and SB-415286. J Cell Mol Med 2009; 13:9B 3906 - 17; http://dx.doi.org/10.1111/j.1582-4934.2008.00389.x; PMID: 18624766
- Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta 1992; 1114:147 - 62; PMID: 1333807
- Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract 2007; 77:Suppl 1 S49 - 57; http://dx.doi.org/10.1016/j.diabres.2007.01.033; PMID: 17478001
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 2008; 104:1433 - 9; http://dx.doi.org/10.1111/j.1471-4159.2007.05194.x; PMID: 18088381
- Raina AK, Monteiro MJ, McShea A, Smith MA. The role of cell cycle-mediated events in Alzheimer’s disease. Int J Exp Pathol 1999; 80:71 - 6; http://dx.doi.org/10.1046/j.1365-2613.1999.00106.x; PMID: 10469261
- Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci 1997; 17:3588 - 98; PMID: 9133382
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 1996; 68:850 - 8; http://dx.doi.org/10.1021/ac950914h; PMID: 8779443
- Hoffmann R, Metzger S, Spengler B, Otvos L Jr.. Sequencing of peptides phosphorylated on serines and threonines by post-source decay in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Mass Spectrom 1999; 34:1195 - 204; http://dx.doi.org/10.1002/(SICI)1096-9888(199911)34:11<1195::AID-JMS881>3.0.CO;2-C; PMID: 10548813
- Lu PJ, Zhou XZ, Liou YC, Noel JP, Lu KP. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J Biol Chem 2002; 277:2381 - 4; http://dx.doi.org/10.1074/jbc.C100228200; PMID: 11723108