Abstract
CDK8 is either amplified or mutated in a variety of human cancers, and CDK8 functions as an oncoprotein in melanoma and colorectal cancers. Previously, we reported that loss or reduction of CDK8 results in aberrant fat accumulation in Drosophila and mammals, suggesting that CDK8 plays an important role in inhibiting lipogenesis. Epidemiological studies have identified obesity and overweight as the major risk factors of endometrial cancer, thus we examined whether CDK8 regulates endometrial cancer cell growth by using several endometrial cancer cell lines, including KLE, which express low levels of CDK8, as well as AN3 CA and HEC-1A cells, which have high levels of endogenous CDK8. We observed that ectopic expression of CDK8 in KLE cells inhibited cell proliferation and potently blocked tumor growth in an in vivo mouse model. In addition, gain of CDK8 in KLE cells blocked cell migration and invasion in transwell, wound healing and persistence of migratory directionality assays. Conversely, we observed the opposite effects in all of the aforementioned assays when CDK8 was depleted in AN3 CA cells. Similar to AN3 CA cells, depletion of CDK8 in HEC-1A cells strongly enhanced cell migration in transwell assays, while overexpression of CDK8 in HEC-1A cells blocked cell migration. Furthermore, gene profiling of KLE cells overexpressing CDK8 revealed genes whose protein products are involved in lipid metabolism, cell cycle and cell movement pathways. Finally, depletion of CDK8 increased the expression of lipogenic genes in endometrial cancer cells. Taken together, these results show a reverse correlation between CDK8 levels and several key features of the endometrial cancer cells, including cell proliferation, migration and invasion as well as tumor formation in vivo. Therefore, in contrast to the oncogenic effects of CDK8 in melanoma and colorectal cancers, our results suggest that CDK8 plays a tumor-suppressive role in endometrial cancers.
Introduction
Cyclin-dependent kinases (CDKs) play fundamental roles in regulating cell proliferation and differentiation in eukaryotes.Citation1-Citation3 Dysregulation of CDKs and their regulatory partners, known as cyclins, disrupts proper control of cell proliferation, differentiation or apoptosis, thereby leading to abnormal development and diseases such as cancer. There are 21 CDK family members in mammals.Citation1-Citation3 Many studies in the past three decades have provided significant insights into understanding the roles of several CDKs, such as CDK1, CDK2, CDK4 and CDK6, in regulating cell proliferation and how dysregulation of these CDKs and their regulatory cyclins contributes to tumorigenesis.Citation2,Citation4,Citation5 However, much less is known about the potential roles of other CDKs and their deregulation in human cancers and other diseases.
For the following reasons, we focus our study on functions of CDK8 in cancer cells: first, recent studies have linked dysregulation of CDK8 to a number of human cancers, and CDK8 is identified as an oncoprotein,Citation6,Citation7 yet the underlying mechanisms of how elevated CDK8 promotes tumorigenesis remain unclear. Second, because CDK8 is a kinase, there is considerable interest in developing cancer drugs by targeting CDK8.Citation8 However, a better understanding of CDK8 functions in vivo is required to evaluate the efficacy and to avoid potential side effects of this approach. Third, biochemical studies have revealed that CDK8 is the only enzymatic subunit of the transcription cofactor complex called Mediator complex, which bridges specific transcription factors and the general transcription machinery.Citation9-Citation13 Mediator complex is composed of up to 30 different subunits, most of which are well conserved in eukaryotes.Citation10,Citation14,Citation15 It is proposed that the Mediator complex is involved in most, if not all, of the RNA polymerase II-mediated transcription.Citation10 Despite many biochemical studies, the in vivo function and regulation of CDK8 and other Mediator subunits in multicellular organisms are still poorly understood.Citation9,Citation16,Citation17
The importance of CDK8 in maintaining tissue homeostasis in multicellular organisms is highlighted by accumulating clinical evidence that links dysregulated CDK8 to a variety of human cancers in recent years.Citation17 For example, CDK8 gene is amplified or overexpressed in colorectal and gastric cancers,Citation6,Citation17-Citation23 and overexpression of wild-type CDK8 in untransformed 3T3 cells led to anchorage-independent growth.Citation6 In addition, loss of the histone variant macroH2A increases the expression of CDK8 in melanoma.Citation7 Importantly, knocking down CDK8 effectively blocks the proliferation of melanoma and colon cancer cells,Citation6-Citation8,Citation24 suggesting that gain of CDK8 is an important oncogenic determinant. An oncogenic role of CDK8 is also supported by studies of walleye dermal sarcoma, which is caused by walleye dermal sarcoma virus. About 30% of adult walleye can develop this type of hyper-proliferative skin disease, and a retroviral cyclin (rv-cyclin) encoded by the walleye dermal sarcoma virus was shown to bind to and potentiate the kinase activity of CDK8, thereby causing the skin cancer in walleye.Citation25,Citation26 Taken together, these studies suggest that CDK8 can function as an oncoprotein, and elevated CDK8 provides a proliferative advantage for certain type of cancers.
CDK8, however, is not always amplified or overexpressed in other types of cancer. For example, the genomic region that harbors CDK8 gene (13q.12) is frequently deleted in esophageal squamous cell carcinoma, bladder cancer, breast cancer and lung adenocarcinoma.Citation17,Citation27-Citation29 In addition, two missense CDK8 point mutations, D189N and R424C, have been identified in colorectal adenocarcinoma and pulmonary neuroendocrine carcinoma, respectively,Citation30 and our bioinformatic analyses indicate that the D189N point mutant is likely to abrogate the CDK8 kinase activity.Citation17 Although it is still unclear why both gain and loss of CDK8 can contribute to tumorigenesis, these studies demonstrate that CDK8 plays a critical role in maintaining tissue homeostasis.Citation17,Citation31 A better understanding of the function and regulation of CDK8 during development and the role of CDK8 in different types of cancers are essential to understand how dysregulation of CDK8 contributes to tumorigenesis.
Recently, we found that CDK8 plays a conserved role in inhibiting lipogenesis, and loss or reduction of CDK8 leads to aberrant fat accumulation in Drosophila and mammals.Citation31 Epidemiological studies have established a positive correlation between obesity and certain types of cancers, particularly breast and endometrial cancers.Citation32-Citation35 However, whether CDK8 plays a role in endometrial cancer has not been explored to date. In this report, we present our functional analysis of CDK8 in endometrial cancer cells. Using several endometrial cancer cell lines, we observed that ectopic expression of CDK8 inhibits, while depletion of CDK8 enhances, several features of the endometrial cancer cells, including cell proliferation, migration and invasion. Importantly, gain of CDK8 in endometrial cancer cells potently blocks tumor initiation or growth in an in vivo mouse model, while depletion of CDK8 enhances tumor growth. Taken together, these observations suggest a tumor-suppressive role of CDK8 in endometrial cancer cells.
Results
Overexpression of CDK8 inhibits growth of human endometrial cancer KLE cells
To investigate potential roles of CDK8 in regulating endometrial cancer growth, we analyzed the protein levels of CDK8 by western blot in five commonly used endometrial cancer cell lines: AN3 CA, ECC-1, HEC-1A, KLE and RL95-2 cells. We found that CDK8 protein was relatively low in KLE cells, while AN3 CA and HEC-1A cells had the highest CDK8 protein expression among all cells examined (). To determine the functional significance of varied CDK8 levels in endometrial cancer cells, we focused our analyses on KLE and AN3 CA cells. First, we tested whether ectopic expression of CDK8 in KLE cells, which have low CDK8 levels, had any effect on cell growth. Specifically, we used a retroviral expression vector encoding wild-type CDK8 and established a stable KLE cell line with gain of CDK8. Compared with the control, which was transduced with the empty vector, this KLE cell line had increased CDK8 expression as assayed by quantitative reverse transcription polymerase chain reaction (qRT-PCR, ; primers are listed in Table S1) and CDK8 protein levels by western blot ().
Figure 1. Overexpression of CDK8 abrogates tumorigenesis in KLE cells. KLE cells were stably transduced with retroviral expression vector encoding CDK8 and the empty vector control. (A) The levels of CDK8 and CycC in five endometrial cancer cell lines analyzed by western blots. (B) qRT-PCR analysis of relative mRNA level of CDK8 and (C) western blot analysis of CDK8 protein levels were shown compared with empty vector control after puromycin selection. (D) Growth curve of CDK8 overexpressing KLE cells. Cells (2.0 × 105 per well) were seeded in 6-well plate, and the number of cells per well was shown for 5 d (n = 3). (E) The effects of CDK8 overexpression on cell cycle progression of the KLE cells analyzed by flow cytometry analysis. (F) Tumor growth of CDK8 overexpression or control KLE cells in 6–8-week-old female nude mice (n > 14 separate mice in each group). Cells (2.0 × 105) were implanted in two flanks of each mouse, and the volume of tumors was assessed in indicated days. (G) Tumor weight was measured at the completion of the experiment. The data shown as mean ± SEM from triplicates calculated using two-factor repeated measure analysis of variance followed by Fisher’s last significant different test for multiple comparisons.
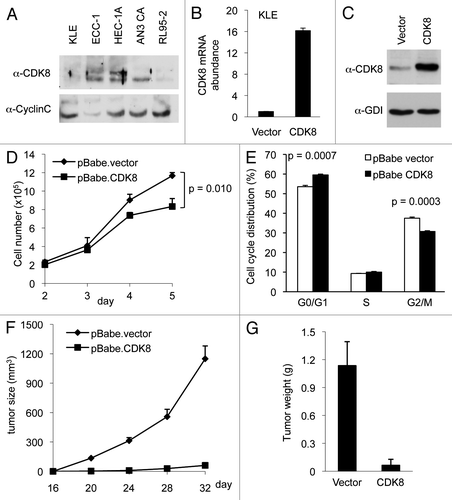
Next, we analyzed whether ectopic expression of CDK8 could affect the growth of KLE cells. As shown in , we observed that CDK8 overexpression reduced the growth of KLE cells in cell culture. To examine the effect of CDK8 overexpression on cell cycle distribution, we performed flow cytometry analyses and observed that gain of CDK8 in KLE cells increased the population of cells in G1 phase and decreased that in G2/M phase (). This result is consistent with the observation that CDK8 affects G1/S-phase transition by inhibiting the activity of E2F1.Citation36,Citation37 Furthermore, to examine the effect of increased CDK8 on tumor growth in vivo, we used a nude mouse model and implanted KLE cells by subcutaneous injection. The tumor sizes were measured every 4 d started from day 16 after implantation. We observed that ectopic expression of CDK8 in KLE cells significantly blocked the tumor initiation in vivo (), resulting in reduced tumor size and weight (). Taken together, these data suggest that gain of CDK8 potently inhibits the growth of endometrial cancer cells.
Depletion of CDK8 enhances the growth of AN3 CA cells
We reasoned that if CDK8 negatively regulates endometrial cancer cell growth, as we observed in KLE cells (), then we would expect opposite effects when CDK8 is depleted in AN3 CA cells, which have high CDK8 levels. Thus to further examine the notion that CDK8 inhibits the growth of endometrial cancer cells, we depleted CDK8 in AN3 CA cells by transducing the cells with pLKO.1-shRNAs, which were previously shown to specifically target CDK8.Citation6,Citation7,Citation31,Citation36,Citation38 Similar construct targeting GFP (sh-GFP) was used as the negative control. Transduced cells were selected by puromycin for over 2 weeks, and the efficiency of CDK8 knockdown was verified by qRT-PCR and western blot. Indeed, we observed that the levels of CDK8 mRNA () and CDK8 protein () were significantly reduced by sh-CDK8-2.
Figure 2. Depleting CDK8 promotes tumorigenesis in AN3 CA cells. AN3 CA cells were transduced with pLKO.1-shRNA targeting CDK8 or with control sequence targeting GFP. (A) The relative mRNA level of CDK8 was analyzed by qRT-PCR analysis, and (B) the CDK8 protein levels were analyzed by western blot. (C) AN3 CA cells or control cells (1.5 × 105 per well) were seeded in 6-well plate, and the number of cells per well was shown for 5 d (n = 4). (D) The effects of CDK8 reduction on cell cycle progression of the AN3 CA cells analyzed by flow cytometry analysis. (E) Colony formation experiments were conducted in 6-cm cell culture dish. 1,000 cells were seeded and allowed to grow for 2 weeks, and then the cells were stained with crystal violet (left). (F) The size and number of colonies were quantified by Fuji (ImageJ) software. (G) Tumor growth of CDK8 knockdown or control AN3 CA stable cells in 6–8-week-old female nude mice (n > 14 separate mice in each group). Cells (2 × 106) were implanted in two flanks of each mice and the volume of tumors were assessed in indicated days.
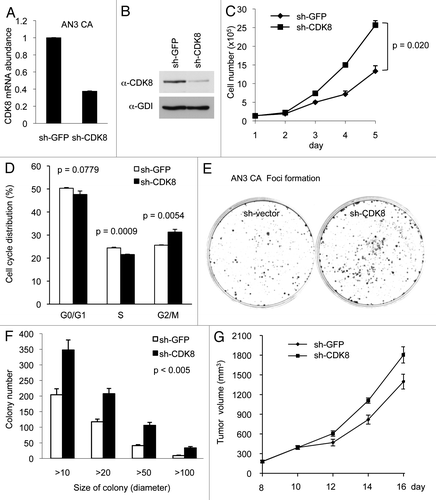
Using these cells, we then analyzed the effect of CDK8 depletion on cell growth. As shown in , depletion of CDK8 increased the proliferation of AN3 CA cells, and this difference was especially significant at day 4 and day 5. In addition, we tested the effect of CDK8 reduction on cell cycle distribution by flow cytometry analyses, and we observed that knockdown of CDK8 in AN3 CA cells reduced population of cells in S phase and increases the G2/M phase (). These observations suggest that high levels of CDK8 may negatively regulate the proliferation of AN3 CA cells. Next, we tested whether CDK8 levels affect tumorigenic potential of cells by foci formation assay. As shown in , depletion of CDK8 in AN3 CA cells increased foci formation. This effect of CDK8 on the number of foci in different range of foci size was highly significant, as quantified in . Furthermore, we examined whether reduction of CDK8 enhanced the growth of AN3 CA cells in vivo by using a nude mouse model. After 2 wk of implantation, we observed that the tumor sizes were about 20% larger in AN3 CA cells with reduced CDK8, and this difference is statistically significant (). Therefore, the results summarized in suggest that CDK8 negatively regulates the growth of AN3 CA cells, and reduction of CDK8 provides growth advantage of AN3 CA cells in vivo. These results are consistent to what we observed in KLE cells ().
Identification of signaling pathways regulated by CDK8
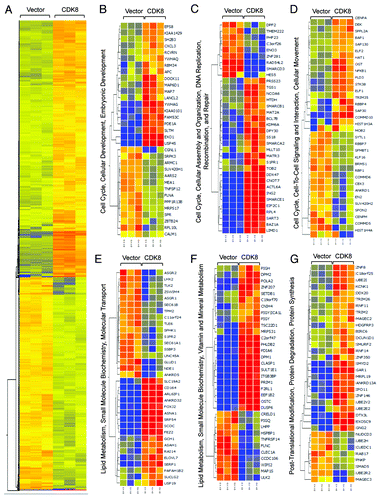
CDK8 is best known to regulate RNA polymerase II-dependent gene expression by phosphorylating certain transactivators during transcriptional activation.Citation9,Citation16,Citation17,Citation39 To understand the molecular mechanisms by which CDK8 represses endometrial tumor growth at high resolution, we performed genome-wide analyses to identify the genes whose expression was regulated by CDK8. KLE cells with low endogenous CDK8 protein expression were transduced with retroviral plasmid encoding wild-type CDK8. We then compared gene expression profiles in cells expressing CDK8 and control by microarray analysis. We identified 2,128 transcripts that were upregulated, and 1,725 genes that were downregulated by CDK8 expression (a change in expression of more than 1.5-fold and threshold of false discovery rate less than 0.05) (Table S2; ). Consistent with the known functions of CDK8, we found that genes whose products are involved in regulating cell cycle, cellular assembly, cell-to-cell signaling, cell movement and cellular and embryonic development pathways were altered by gain of CDK8 (). In addition, lipid metabolism, small molecule biochemistry and metabolism pathways were also affected by gain of CDK8 (). The observation of CDK8-regulated genes whose products are involved in lipid metabolism further supports a role of CDK8 in lipogenesis as we recently reported.Citation31 Finally, we also observed genes whose products are involved in regulating post-translational modification and protein degradation (). These microarray data were verified in part by analyzing a few genes from these lists using qRT-PCR assay (Fig. S1). Further analyses are required to elucidate how gain of CDK8 alters the expression of these genes belonging to distinct functional categories. Because metastasis and aberrant lipid metabolism are critical features during tumorigenesis, we further analyzed the role of CDK8 in regulating cell migration and lipid metabolism.
Figure 3. Genome-wide analysis of CDK8-regulated transcriptome. (A) Treeview of genes that are differentially regulated by CDK8. Red color indicates upregulated genes, while blue color indicates downregulated genes. (B–G) Ingenuity Pathway Analysis illustrates the differentially regulated gene networks and biological pathways following CDK8 overexpression.
CDK8 inhibits cellular migration and invasion of endometrial cancer cells with low CDK8
Our analyses using KLE and AN3 CA cells suggest that CDK8 negatively regulates the growth of endometrial cancer cells. Since metastasis is a critical step during cancer progression, and our microarray analyses suggest that CDK8 may regulate the expression of genes whose products are involved in regulating cell migration (), we asked whether CDK8 plays a role in regulating the migration of endometrial cancer cells. First, we tested whether gain of CDK8 in CDK8-low KLE cells had any effect on cell migration and invasion.
To monitor cell migration, transwell assays were performed as described in details previously.Citation53 Compared with the vector control, we observed that KLE cells stably expressing CDK8 significantly reduced the number of cells in the lower chambers (), suggesting an inhibitory effect of CDK8 on cell migration. Next, we tested whether CDK8 regulated cancer cell invasion, by using three-dimensional invasiveness assays. Ectopic expression of CDK8 significantly reduced the depth of cells penetrating to the collagen (), suggesting that increased CDK8 hampers the cancer cell invasion. Together, these observations demonstrate that gain of CDK8 suppresses endometrial cancer cell migration and invasion, which is consistent with a tumor-suppressive role of CDK8 in endometrial cancer.
Figure 4. Effects of CDK8 on cell migration and invasion in endometrial cancer cells with low levels of CDK8. KLE cells stably overexpressing CDK8 were analyzed for cell migration by transwell assay and for invasion by three-dimensional invasiveness assay. (A) Crystal violet dye staining of cells that migrated through the 8-μm pore in the transwell assays. The data (right) were shown as mean ± SEM of the number of cells migrated from three separate experiments. (B) PI staining of invasive cells by three-dimensional invasiveness assay was shown. The data were shown as mean ± SEM of the z-sections of invasive cells from three separate experiments. (C) Cell movement and quantification of the velocity for wound healing and PMD in CDK8 overexpressing KLE cells or control cells were presented.
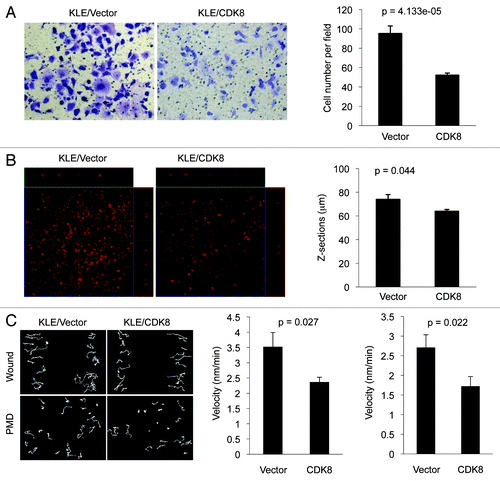
To further examine the role of CDK8 in regulating endometrial cancer cell migration, we performed wound healing and persistence of migratory directionality (PMD) assays, since the speed of wound closure is the combination of cell migratory velocity and PMD. KLE cells with overexpressed CDK8 were cultured to reach 100% confluence, the cell monolayer was scratched generate a wound and cell movements toward the “wound” were documented with time-lapse video. Tracks of cell movement were projected, and the velocity of cell movement was calculated. Representative examples of cell movements for wound healing and PMD in CDK8-overexpressing KLE cells or control cells were shown in (left panel). We observed that ectopic expression of CDK8 in KLE cells significantly reduced the velocity of cell movements in both wound healing (, middle) and PMD assays (, right panel), suggesting that increased CDK8 negatively regulates KLE cell movements.
Effects of CDK8 in cellular migration and invasion in endometrial cancer cells with high levels of CDK8
To further analyze the inhibitory role of CDK8 in cellular migration and invasion, we performed similar assays using the endometrial cancer cells with relatively high levels of endogenous CDK8 expression, such as AN3 CA and HEC-1A cells (). First, we examined whether reducing CDK8 could enhance migratory ability of AN3 CA cells originally derived from lymph node metastasis. Representative examples of cell migration in PMD assay were shown in . Compared with the controls, we found that depleting CDK8 in AN3 CA cells significantly increased the velocity of cell movements (, right panel), consistent with the observation using KLE cells ().
Figure 5. Effects of CDK8 on cell migration and invasion in endometrial cancers with high levels of CDK8. (A) PMD analysis was conducted in endometrial cancer AN3 CA cells following treatment with shRNA targeting CDK8 and control. The velocity was determined for individual cells as indicated (right). Data was presented as mean ± SEM of n > 20 separate event. (B) Western blot was performed to determine the knockdown efficiency of shRNA targeting CDK8 in AN3 CA cells. (C) AN3 CA cells with knockdown of CDK8 by shRNA were analyzed for cell migration by transwell assay. (D) western blot was performed to determine the knockdown efficiency of shRNA targeting CDK8 in HEC-1A cells. (E) HEC-1A cells with knockdown of CDK8 by shRNA were analyzed for cell migration by transwell assay. (F) Transwell migration arrays were conducted of HEC-1A cells transduced with retroviral vector encoding CDK8. Transwell data was presented as mean ± SEM of n > 9 random microscopic field.
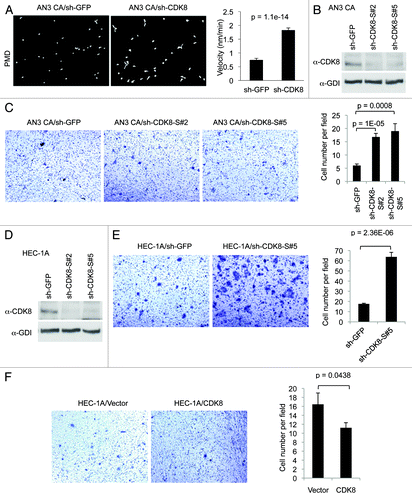
Because shRNA approach to deplete CDK8 could generate false-positive results due to potential off-target effects of RNA interference, we tested an independent set of shRNAs consisting of five novel shRNA constructs targeting hCDK8, designated as “sh-CDK8-S#1” to “sh-CDK8-S#5.” We found two of them, sh-CDK8-S#2 and sh-CDK8-S#5, effectively reduced CDK8 in AN3 CA cells () and HEC-1A cells (). We then performed transwell assay, and found that depletion of CDK8 with either of these sh-CDK8 constructs significantly enhanced cell migration in this assay (). We observed similar effect of CDK8 depletion in another endometrial cancer cell line, HEC-1A, which expresses high levels of CDK8 (). Furthermore, we tested whether further increased CDK8 in HEC-1A cells has effects on cellular migration using the transwell assay. As shown in , overexpression of wild-type CDK8 inhibits HEC-1A cell migration. Taken together, these data obtained from both AN3 CA and HEC-1A cells and additional sh-CDK8 constructs further confirmed that CDK8 inhibits the migration of endometrial cancer cells.
CDK8 inhibits lipogenesis in endometrial cancer cells
For the following reasons, we sought to examine whether CDK8 affected the endometrial cancer growth by regulating the lipogenic gene expression. First, we recently showed that CDK8 inhibited sterol regulatory element-binding protein (SREBP-1)-dependent lipid production in Drosophila and mammalian hepatic cells.Citation31 SREBPs are a family of master transcription factors that controls the expression of lipogenic enzymes.Citation40-Citation43 Second, our microarray analysis revealed altered expression of genes whose products were involved in regulating lipid metabolism when CDK8 is overexpressed (). Third, aberrant lipid metabolism is a universal feature of a variety of human cancer cells.Citation44
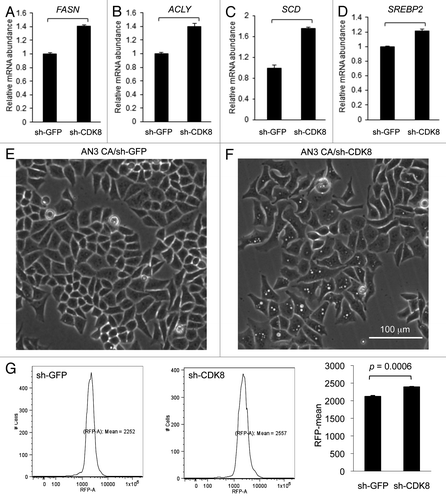
To test whether CDK8 is required for lipogenic gene expression in cancer cells, we analyzed the effect of CDK8 depletion in AN3 CA cells on mRNA level of SREBP-1 and SREBP-2 target genes. We observed that depletion of CDK8 in AN3 CA cells by shRNA (sh-CDK8-2) significantly increased expression of several well-characterized SREBP-1 target genes, such as fatty acid synthase (FASN, ), ATP citrate lyase (ACLY, ) and stearoyl-CoA desaturase-1 (SCD1, ). In addition, we observed significantly increased expression of SREBP-2 (), but not SREBP-1A/1C (data not shown). These observations are consistent with our prior report that CDK8 inhibits the activity of nuclear SREBP in Drosophila and mammalian hepatic cells.Citation31 The effects of CDK8 depletion on lipid accumulation were visualized under phase contrast microscopy; note the lipid droplets in cells shown in , compared with the control (). Accumulated lipid was stained with Nile Red and quantified with flow cytometry (). To support continuous cell proliferation, cancer cells need de novo synthesis of fatty acids and phospholipids to provide membrane materials, which presumably explains how lipid metabolism is coupled with cancer cell proliferation, and how targeting lipogenic pathways could stop tumor growth.
Figure 6. Depleting CDK8 enhances lipid accumulation in endometrial cancer AN3 CA cells. (A–D) Quantification of gene expression by using qRT-PCR. (E–F) The phase contrast images were acquired, showing lipid droplet accumulation in cells with depleted CDK8 (F) compared with the control (E). (G) Cells were stained with Nile Red and subjected to flow cytometry assays to quantify the lipid levels.
Discussion
Several lines of evidence suggest that CDK8 functions as an oncoprotein in certain type of cancers. CDK8 serves as an oncoprotein in colorectal cancer by promoting colon cancer cell proliferation, β-catenin-driven transformation and β-catenin activated gene expression.Citation6,Citation36 There are also reports, however, demonstrating loss of CDK8 in other cancer types.Citation17 In this study, we observed that introduction of CDK8 in endometrial cancer cells that have low levels of CDK8 (KLE cells) blocks cell proliferation, tumor growth, cellular migration and invasion (, and ). Conversely, depletion of CDK8 in endometrial cancer cells that have high levels of CDK8 (AN3 CA and HEC-1A cells) enhances tumor growth and cellular migration and invasion ( and ). In addition, reduction of CDK8 upregulates the expression of lipogenic genes in endometrial cancer cells, supporting our recent observation that CDK8 negatively regulates lipogenesis ( and ) through targeting SREBP-1.Citation31 Thus, in contrast to its oncogenic role reported in recent years, the current study revealed an unexpected function of CDK8 in suppressing the growth of endometrial cancer cells ( and ).
How do we reconcile these seemingly contradictory roles (oncogenic vs. tumor-suppressive functions) of CDK8 during tumorigenesis in different cancer types? We would like to propose that at least part of the answer lies in the fact that function of CDK8 is context-dependent. CDK8 was biochemically identified as a subunit of the CDK8 submodule, which contains CDK8, Cyclin C (CycC), MED12 and MED13; thus, CDK8 can function within the CDK8 submodule. However, only a fraction of CDK8 binds to MED12 and MED13 in mammalian cells, suggesting that CDK8 can also have Mediator-independent functions.Citation45-Citation47 In addition, although CDK8 module is generally thought to inhibit transcription, the CDK8 kinase activity is not always required for this function for different transactivators.Citation9 Therefore, the function of CDK8 can be defined as the CDK8 kinase or the transcription function of the CDK8 submodule.Citation17 Tumorigenic effects caused by gain-of-function mutations define oncoproteins, while tumorigenic effects caused by loss-of-function mutations define tumor suppressors. CDK8 amplification, deletion or mutations can have different consequences on the activities of the CDK8 kinase or the transcription function of the CDK8 submodule: amplification of CDK8 can increase CDK8 kinase activity, but may form partial CDK8 submodules, thus generating dominant-negative effects on CDK8 submodule functions. In contrast, a CDK8 point mutation may abrogate CDK8 kinase activity, but has little effect on the organization of CDK8 submodule, thus may not affect the CDK8 submodule functions that are independent of CDK8 kinase activity. Furthermore, deletion of CDK8 compromises both CDK8 kinase activity and the function of the CDK8 submodule. These three scenarios suggest that it will be important to clarify the functional consequences of CDK8 amplification, deletion or mutations in different cancers in future studies.
These opposite effects can affect tissue-specific transactivators differently. CDK8 is known to directly regulate the activities of several transcription factors in metazoans, including p53, E2F1, β-catenin, SMADs and Notch intracellular domain (NICD),Citation6,Citation16,Citation17,Citation36,Citation48-Citation50 all of which are commonly dysregulated in a wide variety of human cancers. We recently showed that CDK8 inhibits lipogenesis by directly phosphorylating SREBP and promoting the degradation of SREBP in Drosophila and mammalian cells.Citation31 Interestingly, CDK8-dependent phosphorylation positively regulates the activity of p53 and SMADs but negatively regulates the activity of E2F1, NICD and SREBP.Citation6,Citation16,Citation17,Citation31,Citation36,Citation48-Citation50 Thus, it is reasonable to postulate that CDK8 amplification or mutation can differentially affect these transactivators in a tissue-specific or biological context-specific manner, which may explain why CDK8 amplification or mutation can be oncogenic or tumor-suppressive in different tissues. Clearly, either gain or loss of CDK8 can contribute to tumorigenesis, and it would be an oversimplification to designate CDK8 as either an oncoprotein or a tumor suppressor.
The results summarized in this report raise several important questions that require further investigations in the future. First, we note that our results are not sufficient to define CDK8 as a “tumor suppressor gene” in endometrial cancer. To our knowledge, the status of CDK8 in endometrial cancer patient samples has not been examined to date. Second, it will be important to analyze whether CDK8 levels or activities are altered in endometrial cancer patients, and whether low levels of CDK8 expression or activity correlate with poor prognosis. Interestingly, aberrant activation of the canonical Wnt/β-catenin signaling pathway was proposed to contribute to neoplastic transformation of the endometrium.Citation51 To rigorously examine the relationships among CDK8, β-catenin and estrogen receptor in endometrial cancer, it will be necessary to simultaneously analyze the status of these factors in the same set of endometrial cancer patient samples. Third, although our data suggest that the impact of CDK8 on cell cycle ( and ) and apoptosis (Fig. S2) are statistically significant, such effects seem rather marginal, and the molecular mechanisms by which CDK8 inhibits endometrial cancer initiation or growth are still not known. Finally, in our experiments using pBabe-CDK8 to overexpress wild-type CDK8 in KLE cells ( and ), it is not known whether the levels of CDK8 are comparable to the physiological level of CDK8, which has not been documented in normal human endometrial epithelium. If the level of overexpressed CDK8 is higher than physiological levels of CDK8, then the results based on this approach may not be physiologically relevant but may have implications to pathophysiological effect of CDK8 overexpression in endometrial cancer cells.
Our observations on the role of CDK8 in endometrial cancer cells may have important clinical implications; our results raise the potential concerns about approaches to inhibit CDK8 kinase activity in treating cancers, and additional studies are necessary to evaluate the potential side effects of CDK8 inhibition. Nevertheless, developing CDK8-specific inhibitor is still an attractive approach to treat cancers that display gained CDK8, considering that such CDK8-specific inhibitors may benefit a substantial portion of melanoma and colorectal cancer patients.
Materials and Methods
Plasmids and antibodies
The shRNA constructs that specifically target human CDK8 (pLKO-sh-CDK8-1 and pLKO-sh-CDK8-2) were generously provided by Dr Ron Firestein.Citation6 These constructs have been shown to specifically knockdown CDK8 previously.Citation6,Citation7,Citation31,Citation36,Citation38 We observed that pLKO-sh-CDK8-2 works more efficiently in depleting CDK8 than pLKO-sh-CDK8-1 in AN3 CA cells, thus pLKO-sh-CDK8-2 was used for further analysis in this work. Besides these two sh-CDK8 constructs, an independent set of sh-CDK8 constructs that target different sequence of hCDK8 mRNA using the same pLKO vector, designated as “sh-CDK8-S#1” to “sh-CDK8-S#5” were purchased from Sigma: sh-CDK8-S#1 (TRCN0000000490), sh-CDK8-S#2 (TRCN0000000491), sh-CDK8-S#3 (TRCN0000194708), sh-CDK8-S#4 (TRCN0000350308) and sh-CDK8-S#5 (TRCN0000350344). Anti-GDI antibody was previously described,Citation52 and anti-CDK8 (D-9) antibody was purchased from Santa Cruz Biotechnology.
Cell culture
Endometrial cancer cell lines (KLE and AN3 CA) were purchased from the American Type Culture Collection (ATCC). KLE cells were cultured in DMEM:F12, AN3 CA cells were cultured in Eagle's minimum essential medium, and the human embryonic kidney 293T cells (HEK293T) were maintained in DMEM. These media were supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin.
Cell transfection and transduction
For transient transfection assay, Superfect Transfection Reagent (Qiagen) was used following the manufacturer’s protocol. For cell transduction, lentiviruses were prepared using Trans-Lentiviral shRNA Packaging Kit (Open Biosystems) following manufacturer’s instruction with modifications. Briefly, lentiviral vector expressing shRNA was introduced into HEK293T cells by transient co-transfection with helper virus with calcium phosphate precipitation. After 6 h, cell culture medium was replaced, and cells were allowed to grow for 36 h to produce viruses. The supernatant was then collected and filtered through a 0.45-µm filter. Cells were infected at approximately 70% confluence in culture medium supplemented with 8 µg/ml polybrene. After 2 d, the medium was changed to basal medium supplemented with 10% FBS and cultured for further assays. Cells were stably selected by supplementing the medium with puromycin (1 µg/ml for KLE cells and 2 µg/ml for AN3 CA cells) for 2 wk. The efficiency for knockdown and overexpression of CDK8 was determined by western blot or qRT-PCR assays.
Cellular proliferation assay and colony formation assays
For cell proliferation assays, cells with stable overexpression or knockdown of CDK8 and controls were seeded at a density of 2.0 × 105 for KLE cells and 1.5 × 105 for AN3 CA cells per well in 6-well cell culture plates. The total number of cells per well was counted for 5 d. For colony formation assays, 1.0 × 103 cells were seeded in 60-mm plates and allowed to grow for 2 wk with the culture medium replaced once every 3 d. The number of colonies formed per plate was stained with crystal violet and quantified by using a Gel-Pro Analyzer (Media Cybernetics, Inc.).
Wound healing and persistence of migratory directionality (PMD) assays
Cells with stable overexpression or knockdown of CDK8 and controls were seeded at the same number per well and cultured in 24-well glass bottom plate (MatTek Corporation) and cultured for 24 h. Cell migration was monitored by using an inverted microscope (Zeiss) at 37°C with 5% CO2. Time-lapse recordings were collected with a charge-coupled-device camera (model 2400) at a 12 min-interval for 24 h, 119 pictures per view were stored and the velocity of cell migration was calculated using the Metamorph software.
For wound healing assay, 100% confluent cell monolayer was scratched using 200 µl tip to draw a linear “wound” and then washed twice with medium to remove debris or the detached cells. Pictures were captured immediately. Cells (n > 20) were counted for migration into the cell-free zone. For PMD assay, 7.0 × 105 of AN3 CA (or HEC-1A) cells transduced with sh-CDK8 or sh-GFP control and 3.0 × 105 of KLE/vector and KLE/CDK8 were seeded in each well. Single cells (n > 30) were calculated. The data of cell motility assay were presented as mean ± SEM from triplicates and statistically evaluated using a one-way analysis of variance (ANOVA).
Transwell migration and three-dimensional invasion assay
Transwell migration and three-dimensional invasion assays were performed as described previously.Citation53
Subcutaneous tumor implantation
KLE or AN3 CA cells (2.0 × 106) cells with either overexpression or knockdown of CDK8 (controls: empty vector or sh-GFP) were implanted by subcutaneous injection in the flank of 6–8-week-old female nude mice. Comparisons were made for more than 14 animals in each group between AN3 CA/sh-GFP and AN3 CA/sh-CDK8, or KLE/vector and KLE/CDK8. The tumor growth rates were examined using serial caliper measurements. The tumor volume were calculated using the equation (a × b2)/2, where “a” and “b” are length and width of the tumor, respectively. At the end of the experiments, tumors were excised and weighed. The differences in tumor volume were logarithm-transformed and statistically analyzed using a linear mixed model. Separate slope and intercepts were computed for each group, then we compared across groups using a global test followed by pair-wise comparisons via linear contrasts. Data prior to day 8 (for AN3 CA cells) or day 16 (for KLE cells) were ignored due to zeroes at day 0 (inability to take logarithms) and an initial nonlinearity or change in some of the animals’ growth patterns prior to day 8 or day 16. Thus, the intercept at day 8 or day 16 is interpretable as initiation of growth, and the slope is interpreted as rate of growth. For the models, we report coefficients, confidence intervals of coefficients and two-sided p-values.
RNA isolation and qRT-PCR
Total RNA was prepared using TRIzol Reagent (Invitrogen) following manufacturer’s instructions. Total RNA (1.0 µg) was subjected to reverse transcription to synthesize cDNA using the SuperScript™ II Reverse Transcriptase Kit (Invitrogen). For qRT-PCR, each reaction (25 μl) consisted 1.0 µl reverse transcription cDNA product and 100 nM of each primer. The primers used for qRT-PCR are listed in Table S1.
Genome-wide gene expression analysis
DNA microarray analysis was performed using the Human Whole Genome OneArray® (Phalanx Biotech Group, Inc.). RNA quality and integrity were determined utilizing an Agilent 2100 Bioanalyzer (Agilent Technologies). Only high quality RNA samples with a RIN of > 7.0 and an A260/280 absorbance ratio of > 1.8 were utilized for further experimentation. RNA was converted to double-stranded cDNA and amplified using in vitro transcription that included aminoallyl UTP, and the aRNA product was subsequently conjugated with Cy5™ NHS ester (GE Healthcare Life Sciences). Fragmented aRNA was hybridized at 42°C overnight using the HybBag mixing system with 1× OneArray Hybridization Buffer (Phalanx Biotech), 0.01 mg/ml sheared salmon sperm DNA (Promega), at a concentration of 0.025 mg/ml labeled target. After hybridization, the arrays were washed according to the OneArray protocol.
Raw intensity signals for each microarray were captured using a Molecular Dynamics™ Axon 4100A scanner, measured using GenePixPro™ Software and stored in GPR format. Data was normalized using quantile normalization with GeneSpring V12.0 software (Agilent). 1.5-fold differentially expressed gene list was generated. The differentially expressed gene list was loaded into Ingenuity Pathway Analysis (IPA) 8.0 software (www.ingenuity.com) to perform biological network and functional analyses.
Additional material
Download Zip (1,003.1 KB)Acknowledgments
This work was supported by a grant from the Pennsylvania Department of Health (C.W.), the Shandong Tai-Shan Scholar Foundation, China (B.K.), the startup funds from Texas A&M Health Science Center and a grant from the American Heart Association (J.Y.J.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24003
References
- Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol 2009; 11:1275 - 6; http://dx.doi.org/10.1038/ncb1109-1275; PMID: 19884882
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153 - 66; http://dx.doi.org/10.1038/nrc2602; PMID: 19238148
- Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997; 13:261 - 91; http://dx.doi.org/10.1146/annurev.cellbio.13.1.261; PMID: 9442875
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005; 30:630 - 41; http://dx.doi.org/10.1016/j.tibs.2005.09.005; PMID: 16236519
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004; 18:2699 - 711; http://dx.doi.org/10.1101/gad.1256504; PMID: 15545627
- Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008; 455:547 - 51; http://dx.doi.org/10.1038/nature07179; PMID: 18794900
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010; 468:1105 - 9; http://dx.doi.org/10.1038/nature09590; PMID: 21179167
- Osherovich L.. CDK8 is enough in colorectal cancer. Science-Business eXchange 2008; 1:5 - 7; http://dx.doi.org/10.1038/scibx.2008.815
- Taatjes DJ. The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci 2010; 35:315 - 22; http://dx.doi.org/10.1016/j.tibs.2010.02.004; PMID: 20299225
- Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci 2005; 30:235 - 9; http://dx.doi.org/10.1016/j.tibs.2005.03.011; PMID: 15896740
- Conaway RC, Conaway JW. Function and regulation of the Mediator complex. Curr Opin Genet Dev 2011; 21:225 - 30; http://dx.doi.org/10.1016/j.gde.2011.01.013; PMID: 21330129
- Conaway RC, Sato S, Tomomori-Sato C, Yao T, Conaway JW. The mammalian Mediator complex and its role in transcriptional regulation. Trends Biochem Sci 2005; 30:250 - 5; http://dx.doi.org/10.1016/j.tibs.2005.03.002; PMID: 15896743
- Malik S, Roeder RG. Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem Sci 2005; 30:256 - 63; http://dx.doi.org/10.1016/j.tibs.2005.03.009; PMID: 15896744
- Bourbon HM, Aguilera A, Ansari AZ, Asturias FJ, Berk AJ, Bjorklund S, et al. A unified nomenclature for protein subunits of mediator complexes linking transcriptional regulators to RNA polymerase II. Mol Cell 2004; 14:553 - 7; http://dx.doi.org/10.1016/j.molcel.2004.05.011; PMID: 15175151
- Bourbon HM. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res 2008; 36:3993 - 4008; http://dx.doi.org/10.1093/nar/gkn349; PMID: 18515835
- Galbraith MD, Donner AJ, Espinosa JM. CDK8: a positive regulator of transcription. Transcription 2010; 1:4 - 12; http://dx.doi.org/10.4161/trns.1.1.12373; PMID: 21327159
- Xu W, Ji JY. Dysregulation of CDK8 and Cyclin C in tumorigenesis. J Genet Genomics 2011; 38:439 - 52; http://dx.doi.org/10.1016/j.jgg.2011.09.002; PMID: 22035865
- Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res 2006; 66:2129 - 37; http://dx.doi.org/10.1158/0008-5472.CAN-05-2569; PMID: 16489013
- Martin ES, Tonon G, Sinha R, Xiao Y, Feng B, Kimmelman AC, et al. Common and distinct genomic events in sporadic colorectal cancer and diverse cancer types. Cancer Res 2007; 67:10736 - 43; http://dx.doi.org/10.1158/0008-5472.CAN-07-2742; PMID: 18006816
- Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, et al. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci USA 2009; 106:7131 - 6; http://dx.doi.org/10.1073/pnas.0902232106; PMID: 19359472
- Seo JO, Han SI, Lim SC. Role of CDK8 and beta-catenin in colorectal adenocarcinoma. Oncol Rep 2010; 24:285 - 91; PMID: 20514474
- Kim MY, Han SI, Lim SC. Roles of cyclin-dependent kinase 8 and β-catenin in the oncogenesis and progression of gastric adenocarcinoma. Int J Oncol 2011; 38:1375 - 83; PMID: 21344156
- Firestein R, Shima K, Nosho K, Irahara N, Baba Y, Bojarski E, et al. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. Int J Cancer 2010; 126:2863 - 73; PMID: 19790197
- Ji JY, Dyson NJ. Interplay between Cyclin-dependent Kinases and E2F-dependent Transcription. In: Enders G, ed. Cell Cycle Deregulation in Cancer: Springer Science 2010:23-41.
- Brewster CD, Birkenheuer CH, Vogt MB, Quackenbush SL, Rovnak J. The retroviral cyclin of walleye dermal sarcoma virus binds cyclin-dependent kinases 3 and 8. Virology 2011; 409:299 - 307; http://dx.doi.org/10.1016/j.virol.2010.10.022; PMID: 21067790
- Rovnak J, Brewster CD, Quackenbush SL. Retroviral cyclin enhances cyclin-dependent kinase-8 activity. J Virol 2012; 86:5742 - 51; http://dx.doi.org/10.1128/JVI.07006-11; PMID: 22379099
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463:899 - 905; http://dx.doi.org/10.1038/nature08822; PMID: 20164920
- Chattopadhyay I, Singh A, Phukan R, Purkayastha J, Kataki A, Mahanta J, et al. Genome-wide analysis of chromosomal alterations in patients with esophageal squamous cell carcinoma exposed to tobacco and betel quid from high-risk area in India. Mutat Res 2010; 696:130 - 8; http://dx.doi.org/10.1016/j.mrgentox.2010.01.001; PMID: 20083228
- Mitra AP, Almal AA, George B, Fry DW, Lenehan PF, Pagliarulo V, et al. The use of genetic programming in the analysis of quantitative gene expression profiles for identification of nodal status in bladder cancer. BMC Cancer 2006; 6:159; http://dx.doi.org/10.1186/1471-2407-6-159; PMID: 16780590
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153 - 8; http://dx.doi.org/10.1038/nature05610; PMID: 17344846
- Zhao X, Feng D, Wang Q, Abdulla A, Xie XJ, Zhou J, et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest 2012; 122:2417 - 27; http://dx.doi.org/10.1172/JCI61462; PMID: 22684109
- Trentham-Dietz A, Newcomb PA, Egan KM, Titus-Ernstoff L, Baron JA, Storer BE, et al. Weight change and risk of postmenopausal breast cancer (United States). Cancer Causes Control 2000; 11:533 - 42; http://dx.doi.org/10.1023/A:1008961931534; PMID: 10880035
- Goodman MT, Hankin JH, Wilkens LR, Lyu LC, McDuffie K, Liu LQ, et al. Diet, body size, physical activity, and the risk of endometrial cancer. Cancer Res 1997; 57:5077 - 85; PMID: 9371506
- Renehan AG, Frystyk J, Flyvbjerg A. Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab 2006; 17:328 - 36; http://dx.doi.org/10.1016/j.tem.2006.08.006; PMID: 16956771
- Webb PM. Commentary: weight gain, weight loss, and endometrial cancer. Int J Epidemiol 2006; 35:166 - 8; http://dx.doi.org/10.1093/ije/dyi301; PMID: 16394116
- Morris EJ, Ji JY, Yang F, Di Stefano L, Herr A, Moon NS, et al. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature 2008; 455:552 - 6; http://dx.doi.org/10.1038/nature07310; PMID: 18794899
- Zhao J, Ramos R, Demma M. CDK8 regulates E2F1 transcriptional activity through S375 phosphorylation. Oncogene 2012; http://dx.doi.org/10.1038/onc.2012.364; PMID: 22945643
- Adler AS, McCleland ML, Truong T, Lau S, Modrusan Z, Soukup TM, et al. CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res 2012; 72:2129 - 39; http://dx.doi.org/10.1158/0008-5472.CAN-11-3886; PMID: 22345154
- Tansey WP. Transcriptional activation: risky business. Genes Dev 2001; 15:1045 - 50; http://dx.doi.org/10.1101/gad.896501; PMID: 11331599
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109:1125 - 31; PMID: 11994399
- Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet 2007; 41:401 - 27; http://dx.doi.org/10.1146/annurev.genet.41.110306.130315; PMID: 17666007
- Goldstein JL, Rawson RB, Brown MS. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem Biophys 2002; 397:139 - 48; http://dx.doi.org/10.1006/abbi.2001.2615; PMID: 11795864
- Raghow R, Yellaturu C, Deng X, Park EA, Elam MB. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab 2008; 19:65 - 73; http://dx.doi.org/10.1016/j.tem.2007.10.009; PMID: 18291668
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7:763 - 77; http://dx.doi.org/10.1038/nrc2222; PMID: 17882277
- Meyer KD, Donner AJ, Knuesel MT, York AG, Espinosa JM, Taatjes DJ. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. EMBO J 2008; 27:1447 - 57; PMID: 18418385
- Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, Taatjes DJ. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol Cell Biol 2009; 29:650 - 61; http://dx.doi.org/10.1128/MCB.00993-08; PMID: 19047373
- Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev 2009; 23:439 - 51; http://dx.doi.org/10.1101/gad.1767009; PMID: 19240132
- Donner AJ, Szostek S, Hoover JM, Espinosa JM. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol Cell 2007; 27:121 - 33; http://dx.doi.org/10.1016/j.molcel.2007.05.026; PMID: 17612495
- Alarcón C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009; 139:757 - 69; http://dx.doi.org/10.1016/j.cell.2009.09.035; PMID: 19914168
- Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell 2004; 16:509 - 20; http://dx.doi.org/10.1016/j.molcel.2004.10.014; PMID: 15546612
- Wang Y, van der Zee M, Fodde R, Blok LJ. Wnt/Β-catenin and sex hormone signaling in endometrial homeostasis and cancer. Oncotarget 2010; 1:674 - 84; PMID: 21317462
- Wang C, Fu M, D’Amico M, Albanese C, Zhou JN, Brownlee M, et al. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol Cell Biol 2001; 21:3057 - 70; http://dx.doi.org/10.1128/MCB.21.9.3057-3070.2001; PMID: 11287611
- Zhou J, Liu Y, Zhang W, Popov VM, Wang M, Pattabiraman N, et al. Transcription elongation regulator 1 is a co-integrator of the cell fate determination factor Dachshund homolog 1. J Biol Chem 2010; 285:40342 - 50; http://dx.doi.org/10.1074/jbc.M110.156141; PMID: 20956529