The metabolic requirements of a proliferating cell differ from those of a cell in homeostasis.Citation1 In order for a cell to duplicate, it must double its genome, protein content and lipid mass. This process requires energy in the form of ATP and NADPH. However, unlike ATP, the amount of NADPH required for biosynthesis is much greater than that needed for homeostasis, which makes the generation of NADPH rate limiting for cellular proliferation. NADPH is used for both macromolecular biosynthesis (e.g., lipids and deoxynucleotide triphosphates) and the maintenance of a reduced intracellular environment.Citation2 Given this dual role, when the demand for NADPH is high (e.g., during proliferation), moderate impacts on NADPH production challenge the maintenance of redox control. As such, the generation of reducing power in the form of NADPH is tightly regulated during proliferation to ensure that a sufficient amount is available to run biosynthetic reactions and protect against oxidative stress.
Recently, we demonstrated that mutant Kras is required for maintenance of established pancreatic tumors, in part through the regulation of anabolic glucose metabolism.Citation3 Mutant Kras drives glucose uptake and its diversion into the non-oxidative arm of the pentose phosphate pathway (PPP) to generate ribose 5-phosphate, which is used in nucleic acid biosynthesis. This was a particularly surprising finding, as such metabolic rewiring bypasses the NADPH-generating oxidative arm of the PPP and suggests that an alternate mechanism for NADPH production must dominate in Kras-transformed pancreatic tumors. In a recent study,Citation4 we used an integrative genetic approach combined with metabolomic tracing experiments to examine this question.
By assessing the role of the two primary anabolic carbon sources (i.e., glucose and glutamine; Gln) on the cellular redox state (a surrogate for NADP+/NADPH) in pancreatic cancers, we found that while both glucose and Gln were required for cell proliferation, only Gln deprivation dramatically increased redox stress. Metabolic rescues of pancreatic cancer cells grown in the absence of Gln revealed that the Gln carbon skeleton (α-ketoglutarate, αKG) was unable to rescue growth unless it was combined with a cocktail of non-essential amino acids (NEAA). These results illustrated an important finding, namely, that pancreatic cancer cells metabolize Gln in a manner that is distinct from the classical αKG-generating mitochondrial pathway that utilizes glutamate dehydrogenase (GLUD1; ).Citation5,Citation6
Figure 1. Gln metabolism is rewired in pancreatic cancer to facilitate NADPH production. (A) Canonical anabolic Gln metabolism. Gln-derived Glu is processed into αKG through mitochondrial GLUD1, which is used for anaplerotic filling of the TCA cycle (green circle). The TCA cycle is coupled to the malate-aspartate shuttle (blue circle), which is used to bring reducing equivalents derived from glycolysis into the mitochondria for oxidative phosphorylation. (B) In pancreatic cancer, Gln metabolism is reprogrammed through the mutant Kras-mediated activation of GOT1 expression and repression of GLUD1. Repression of GLUD1 promotes the mitochondrial aspartate aminotransferase (GOT2)-mediated generation of Asp in the mitochondria. This Asp is released into the cytosol and converted through a series of reactions into pyruvate and reducing potential in the form of NADPH. This series of reactions decouples TCA cycle activity from the malate-aspartate shuttle. Enzymes that facilitate this pathway are presented in upper-case letters. Metabolites are presented in lower-case letters. Cit, citrate; Fum, fumarate; Pyr, pyruvate; Iso, isocitrate; Suc, succinate.
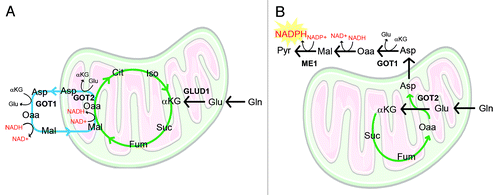
The observation that NEAAs were required downstream of Gln metabolism suggested that transaminases may play a central role in pancreatic cancer. Indeed, we demonstrated that the cytosolic aspartate aminotransferase, GOT1, was required for the maintenance of redox control and for pancreatic cancer cell proliferation. By then tracing Gln metabolism in GOT1 knockdown cells using carbon-13 isotope-labeled Gln and mass spectrometry-based metabolomic profiling,Citation7 it became apparent that Gln metabolism through GOT1 was required for the maintenance of redox balance. Moreover, the altered metabolite distribution in GOT1 knockdown cells suggested that GOT1 functioned upstream of cytosolic malic enzyme (ME1), which we envisioned was required for the generation of reducing equivalents in the form of NADPH (). This model was then validated by knocking down ME1 and again following the distribution of glutamine-derived carbon-13 into downstream metabolites. Subsequent analysis of the oxidized-to-reduced NADP ratio following knockdown of classical NADPH-generating cytosolic enzymes revealed that glucose 6-phosphate dehydrogenase (G6PD, the rate limiting enzyme in the oxidative PPP) or isocitrate dehydrogenase (IDH1) knockdown did not affect the NADP ratio. On the other hand, knockdown of ME1 or GOT1 increased the oxidized-to-reduced NADP ratio, providing clear evidence that this pathway is a major source of NADPH in pancreatic cancers for the maintenance of redox balance.
A consequence of the redox imbalance that occurs by blocking Gln metabolism in pancreatic cancer is the inhibition of proliferation, where suppression of any component enzyme in this pathway impairs growth in a manner similar to that observed upon Gln withdrawal. In fact, this Gln metabolism-mediated redox maintenance is so central to the role of Gln in pancreatic cancer that the defects in proliferation observed upon Gln withdrawal or GOT1 knockdown can be rescued by solely restoring redox balance through media supplementation with a cell-permeable form of reduced glutathione or the antioxidant N-acetyl cysteine. Collectively, these results demonstrate that a principal function of Gln metabolism in pancreatic cancer is to generate reducing power in the form of NADPH, and that this is used, in part, to maintain redox homeostasis, which enables proliferation.
Given the dependence of pancreatic cancer on this Gln metabolism pathway, a major question arises concerning its role in normal cells. Importantly, we demonstrated that GOT1 knockdown did not impair growth across a panel of normal cell lines. Moreover, we found that the signature transforming event in pancreatic cancer, Kras mutation, led to the reprogramming of Gln metabolism. This occurred in part through increasing GOT1 expression and repressing GLUD1 expression. Thus, by changing the ratio of expression of these two enzymes, mutant Kras shunts Gln flux through the aspartate aminotransferase pathway (). The observation that this Gln metabolism pathway is downstream of mutant Kras provides clear rationale as to why pancreatic cancer exhibits this unique metabolic dependency. Finally, in addition to providing several new metabolic therapeutic targets in pancreatic cancer, the findings from this study also suggest that inhibition of Gln metabolism in pancreatic cancer may synergize with therapies that increase ROS, such as chemotherapy and radiation.
Related Research Data
References
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 33; http://dx.doi.org/10.1126/science.1160809; PMID: 19460998
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011; 27:441 - 64; http://dx.doi.org/10.1146/annurev-cellbio-092910-154237; PMID: 21985671
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149:656 - 70; http://dx.doi.org/10.1016/j.cell.2012.01.058; PMID: 22541435
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013; 496:101 - 5; http://dx.doi.org/10.1038/nature12040; PMID: 23535601
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008; 105:18782 - 7; http://dx.doi.org/10.1073/pnas.0810199105; PMID: 19033189
- Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The mTORC1 Pathway Stimulates Glutamine Metabolism and Cell Proliferation by Repressing SIRT4. Cell 2013; 153:840 - 54; http://dx.doi.org/10.1016/j.cell.2013.04.023; PMID: 23663782
- Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 2012; 7:872 - 81; http://dx.doi.org/10.1038/nprot.2012.024; PMID: 22498707