Abstract
Cyclin D1 and its binding partners CDK4/6 are essential regulators of cell cycle progression and are implicated in cancer progression. Our aim was to investigate a potential regulatory role of these proteins in other essential tumor biological characteristics. Using a panel of breast cancer cell lines and primary human breast cancer samples, we have demonstrated the importance of these cell cycle regulators in both migration and stem-like cell activity. siRNA was used to target cyclin D1 and CDK4/6 expression, having opposing effects on both migration and stem-like cell activity dependent upon estrogen receptor (ER) expression. Inhibition of cyclin D1 or CDK4/6 increases or decreases migration and stem-like cell activity in ER−ve (ER-negative) and ER+ve (ER-positive) breast cancer, respectively. Furthermore, overexpressed cyclin D1 caused decreased migration and stem-like cell activity in ER−ve cells while increasing activity in ER+ve breast cancer cells. Treatment of breast cancer cells with inhibitors of cyclin D1 and CDK4/6 (Flavopiridol/PD0332991), currently in clinical trials, mimicked the effects observed with siRNA treatment. Re-expression of ER in two ER−ve cell lines was sufficient to overcome the effects of either siRNA or clinical inhibitors of cyclin D1 and CDK4/6.
In conclusion, cyclin D1 and CDK4/6 have alternate roles in regulation of migration and stem-like cell activity. Furthermore, these effects are highly dependent upon expression of ER. The significance of these results adds to our general understanding of cancer biology but, most importantly, could be used diagnostically to predict treatment response to cell cycle inhibition in breast cancer.
Introduction
Breast cancer is the most common disease in women in the Western world, with 600 000 new patients diagnosed annually, and causes approximately 200 000 deaths each year.Citation1 The introduction of anticancer treatments such as the anti-estrogen tamoxifen have proved effective in improving disease-free survival in breast cancer patients; however, a significant proportion are either resistant to treatment or show disease recurrence.Citation2,Citation3 Recurrences at metastatic sites, in particular lung and bone represent the major cause of mortality in breast cancer patients.Citation4,Citation5 Since the incidence of breast cancer is increasing worldwide, it is essential that we develop more targeted therapies.Citation1
Loss of control of proliferation is considered a hallmark of many cancer types including breast cancer. Proliferation within normal cells is a tightly regulated process; however, cancer may develop when the signals that control proliferation are deregulated. This may occur by a number of mechanisms, including the direct expression of growth factor ligands and receptors by cancer cells or by induction of surrounding normal cells within the tumor stroma to secrete growth factors, which, in turn, allow the uncontrolled proliferation of the cancer cell. In addition, cancer cells are able to proliferate due to the suppression of inhibitors of proliferation. To date numerous tumor suppressor genes have been identified that limit growth and proliferation of many types of cancer.Citation6
Genes involved in cell cycle regulation and treatments aimed to decrease proliferation have therefore been well studied in cancer. Cyclin D1 and its binding partners CDK4/6 are essential components in the cell cycle and regulate the G1 to S-phase transition.Citation7 Cyclin D1 is a known oncogene found to be overexpressed in 25–60% of invasive breast carcinomas with gene amplification found in 10–30% of cases and links to patient prognosis.Citation8-Citation10 Besides its of well-known role in the cell cycle, Cyclin D1 has been implicated in various activities such as chromosomal instability, mitochondrial function, and cellular senescence.Citation11-Citation13 A number of CDKs exist; however, CDK4/6 are thought to be the preferential binding partners of cyclin D1, resulting in modification of Rb phosphorylation,Citation14-Citation16 and have been independently implicated in breast cancer. Reddy et al. showed that CDK4 expression is essential in a Neu-induced breast cancer mouse model,Citation17 and CDK6 has been implicated in the growth of breast cancer cells.Citation18 Most studies have focused on the combined effects of CDK4/6 and potential of inhibiting these proteins to treat breast cancer.Citation19-Citation23
In addition to proliferation, another key feature for breast cancer is migration, thought to be essential in the metastatic process. Tumor cells must possess the ability to migrate and invade into the surrounding tissue in order to leave the primary tumor site; cells then enter the blood stream and lymphatic system by intravasation, followed by extravasation and colonization of surrounding tissue and subsequent formation of metastasis.Citation24 To date, a number of genes have been identified that regulate the migratory process in many cancers, including breast cancer. The best characterized is E-cadherin, a protein which maintains cell-cell adhesion. Loss of this protein causes increased migration and is often downregulated in breast cancer.Citation25,Citation26
Stem cells or cells that possess stem-like cell properties are also thought to be essential in breast cancer initiation and progression. Cells within tumors are heterogeneous and the existence of a small pool of cells, “cancer stem cells” (CSC), are suggested to be responsible for regeneration of tumors.Citation27 CSCs may be identified by cellular markers CD44+/24−, ALDH1 expression, or by mammosphere formation and self-renewal.Citation28,Citation29 MiR-143 and Erk5-dependent upregulation of Cyclin D1 and CDK4 have been reported to drive S-phase progression in mesenchymal rat stem cells, suggesting that targeting cell cycle proteins may deplete tumor cells with stem-like cell characteristics.Citation30 Furthermore cells that possess stem-like properties have been implicated in treatment resistance, emphasizing the need for finding new treatment strategies.Citation29,Citation31,Citation32
We previously have identified an inverse relationship between migration and proliferation in breast cancer using the MDA-MB-231 cell line and identified a novel role of cyclin D1 in the migration of breast cancer cells.Citation33 Given this alternate role of cell cycle regulators, we aimed to extend these observations by widening the selection of cell lines representative of subgroups of breast cancer and primary human breast cancer samples for validation. Furthermore we aimed to assess if cell cycle regulators, in addition to migration, could regulate stem-like cell activity. We have demonstrated using both siRNA and potential clinical inhibitors, cyclin D1, and CDK4/6 affect both migration, mammosphere formation, and ALDH expression, suggesting regulation of stem-like cell activity in addition to migration in breast cancer cell lines and primary samples. Furthermore our data suggests these effects are ER-dependent. These data add significantly to our understanding of the complex co-coordination of key cellular characteristics in different subtypes of cancer, which have implications for the use of anticancer therapies in the clinic.
Results
Knockdown of cell cycle regulators has opposing effects on breast cancer cells dependent upon ER expression
We evaluated the effects of the cell cycle regulators cyclin D1 and CDK4/6 in breast cancer by silencing their gene expression using siRNA. Knockdown of protein expression was confirmed by western blot analysis (). Experiments were performed in two ER−ve breast cancer cell lines (MDA-MB-231 and MDA-MB-468), two ER+ve cell lines (MCF7 and T47D) and 6 primary human breast cancer samples (ER−ve n = 3 and ER+ve n = 3). Inhibition of cyclin D1 expression in ER−ve cell lines significantly increased migration and MS formation, while in ER+ve cell lines a significant decrease was observed (). This was confirmed using the 6 primary human breast cancer samples, 3 expressing ER and 3 lacking ER expression. Silencing of CDK4/6 showed similar effects with increased migration and MS formation in the ER−ve cell line MDA-MB-468 and two ER−ve primary samples tested, while in ER+ve cell lines and primary cells, a decrease was observed. Analyses of the breast cancer stem-like cell marker ALDH following inhibition of cyclin D1 and CDK4/6 were in agreement with the effects observed on mammosphere formation. In breast cancer cell lines, inhibition of cyclin D1 in ER−ve cells caused an increase in ALDH activity, while in ER+ve cells a decrease was observed. Again CDK4/6 siRNA showed similar effects on ALDH activity with a decrease in ER+ve cell lines and an increase in the ER−ve cell line MDA-MB-468 (). Assessment of stem-like activity using two independent methods provides compelling evidence for a genuine effect on stem-like activity.
Figure 1. Downregulation of cyclin D1 and CDK4/6 in breast cancer cell lines and primary human breast cancer cells and effects on migration and mammosphere formation. (A) Immunoblots showing cyclin D1 and CDK4/6 protein expression following siRNA treatment to target cyclin D1, CDK4/6, or non-silencing control. (B) Following siRNA treatment, cells were assessed for migration (upper panel) and mammosphere formation (lower panel) in ER−ve and ER+ve (n = 4) cell lines and primary human breast cancer cells (n = 6). Bar charts represent the mean % number of migrated cells and % mammosphere formation, ± SEM. Cyclin D1 siRNA or CDK4/6 siRNA were compared with control siRNA to generate P values using a two-sided t test assuming equal variance. * Indicates significance, P < 0.05.
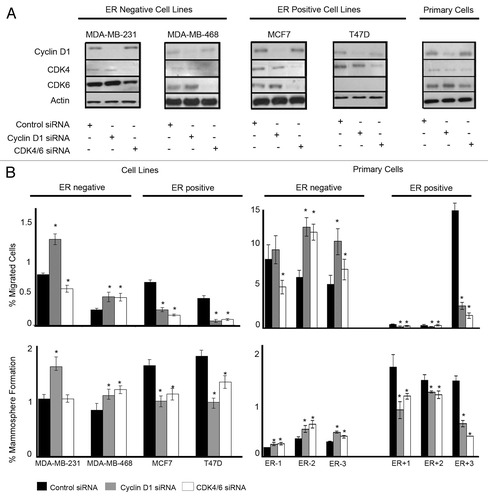
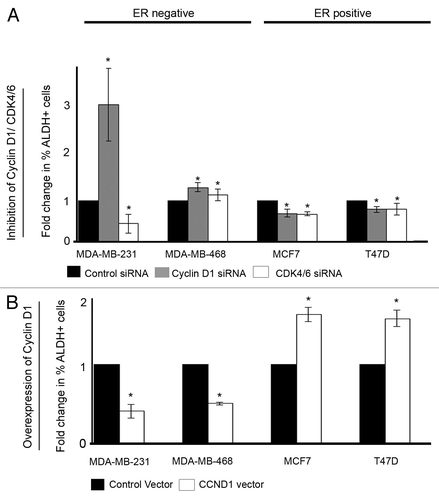
Figure 2. Cell cycle modulation affects ALDH activity. (A) ER−ve and ER+ve cell lines (n = 4) were treated with either control, cyclin D1 siRNA, or CDK4/6 siRNA, and ALDH activity was assessed. Data are presented as mean fold change compared with control siRNA with ± SEM (B) ER−ve and ER+ve cell lines (n = 4) were transfected with either control vector or cyclin D1 vector and ALDH activity assessed. P values were generated using a two-sided t test assuming equal variance. *Indicates significance, P < 0.05.
Overexpression of cyclin D1 protein has opposing effects on breast cancer cells dependent upon ER expression
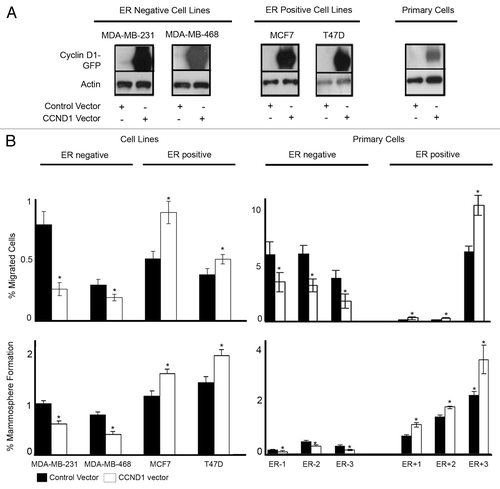
We overexpressed the cyclin D1 protein in 4 breast cancer cell lines and 6 primary breast cancer samples. Overexpression of cyclin D1 was confirmed by western blot analysis (). Overexpression of cyclin D1 caused a significant decrease in both migration and MS formation in ER−ve cell lines and ER−ve primary human breast cancer cells. In ER+ve cells, overexpression of cyclin D1 caused an increase in both migration and MS formation (). Overexpression of cyclin D1 also affected ALDH activity. In ER−ve breast cancer cell lines overexpression of cyclin D1 decreased ALDH activity, while in ER+ve cells ALDH activity was increased ().
Figure 3. Overexpression of cyclin D1 in breast cancer cell lines and primary human breast cancer cells and effects on migration and mammosphere formation. (A) Immunoblots confirming cyclin D1 overexpression following vector transfections. (B) Following vector transfections, cells were assessed for migration (upper panel) and mammosphere formation (lower panel) in ER−ve and ER+ve cell lines (n = 4) and primary human breast cancer cells (n = 6). Bar charts represent the mean % number of migrated cells and % mammospheres formation, ± SEM. Cyclin D1 was compared with control vector to generate P values using a two-sided t test assuming equal variance. *Indicates significance, P < 0.05.
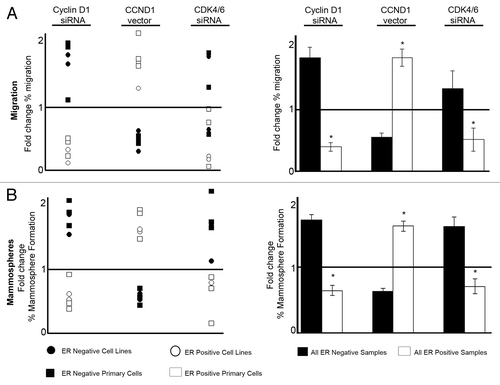
All of the data presented regarding manipulation of cyclin D1 and CDK4/6 for cell lines and primary human breast cancer cells are summarized in . The response of individual breast cancer samples, including cell lines and primary cells to cyclin D1 modulation is clearly determined by the ER expression. The response to CDK4/6 modulation also divides samples according to ER expression with a minority of outliers. Overall, both cyclin D1 and CDK4/6 have ER-dependent effects on migration () and mammosphere formation () of breast cancer cells. Cyclin D1 and CDK4/6 inhibition cause an increase in both migration and mammosphere formation in ER−ve breast cancer cells while having the opposite effect in ER+ve cells. Overexpression of cyclin D1 decreases migration and mammosphere formation in ER−ve breast cancer cells while causing an increase in ER+ve breast cancer cells ().
Figure 4. Summary of effects on cell migration and mammosphere formation resulting from cell cycle modulation in breast cancer lines and primary human breast cancer cells. (A) Summary of migration data plotted as mean fold change compared with corresponding control treatment. Left panel indicates data from both cell lines and primary samples, whereas the right panel summarizes the combined effects on migration according to ER status, with ± SEM (B) Summary of mammosphere data plotted as mean fold change compared with corresponding control treatment. Left panel indicates data from both cell lines and primary samples whereas the right panel summarizes the combined effects on mammosphere formation according to ER status, with ± SEM. Horizontal line at a value of y = 1 indicates no fold change. Controls were compared with each treatment to generate P values using a two-sided t test assuming equal variance. *Indicates significance, P < 0.05.
Inhibitors of cyclin D1 and CDK4/6 undergoing clinical trials have opposing effects on breast cancer cells dependent upon ER expression
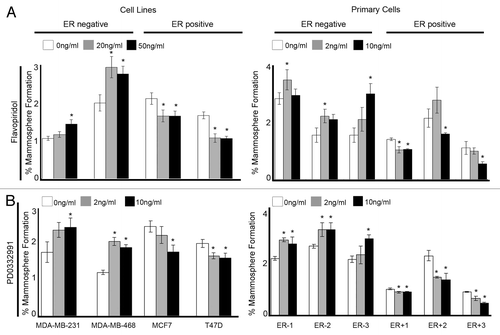
To evaluate the clinical relevance of our findings we selected compounds that are currently undergoing clinical trials to inhibit cyclin D1 and CDK4/6 (Flavopiridol/PD0332991). Breast cancer cell lines and primary human cells were treated in MS culture with a range of concentrations of Flavopiridol and PD0332991. Both drugs significantly increased MS formation in ER−ve cell lines and ER−ve primary human breast cancer samples while in ER+ve cell lines and ER+ve primary human breast cancer samples MS formation were reduced (). These results were consistent with our observations when inhibiting cyclin D1 and CDK4/6 protein expression using siRNA.
Figure 5. Clinical inhibitors of Cyclin D1 and CDK4/6 in breast cancer cell lines and primary human breast cancer cells effects mammosphere formation. Cells were plated into mammosphere culture with the addition of either (A) Flavopiridol or (B) PD0332991 at various concentrations. Bars represent the mean % mammosphere formation for three experiments with ± SEM. Comparisons to 0 ng/ml were made to generate P values using a two-sided t-test assuming equal variance. *Indicates significance, P value < 0.05.
Re-expression of estrogen receptor affects ER-negative cells’ response to cyclin D1 and CDK4/6 modulation
Given the observation that breast cancer cells respond differently to cyclin D1 and CDK4/6 modulation based upon their expression of ER, we evaluated this further. We overexpressed ER in the ER−ve cell line MDA-MD-231, in combination with either cyclin D1 siRNA () or cyclin D1 overexpression (). Where previously we had observed an increase in both migration and MS formation with cyclin D1 inhibition, in combination with overexpression of ER, no increase in migration was observed, and MS formation was decreased (). Conversely, we previously described decreased migration and MS formation with overexpression of cyclin D1 in ER−ve cells; however, in combination with overexpression of ER, we now observed an increase in migration and no change to MS formation ().
Figure 6. Re-expression of ER in ER−ve cell lines reverses the response to cell cycle modulation. MDA-MB-231 breast cancer cells were treated with the indicated siRNA or vector followed by assessment of migration and mammosphere formation. (A) Immunoblot confirmed the silencing of cyclin D1 and overexpression or ER. Bar charts represent the mean % migration and mammosphere formation with ± SEM (B) Immunoblot confirmed the overexpression of GFP-CCND1 and ER. Bar charts represent the mean % migration and mammosphere formation with ± SEM (C) MDA-MB-231 and MDA-MB-468 breast cancer cell lines were transfected with either control or ER vector and plated into mammosphere culture +/− treatment with either Flavopiridol or PD0332991 of various concentrations. P values were generated using a two-sided t test assuming equal variance. *Indicates significance, P < 0.05.
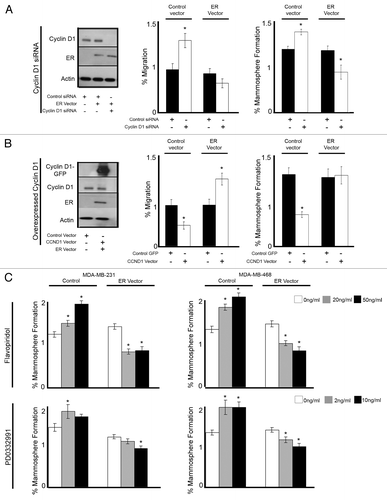
Finally, we treated two ER−ve breast cancer cell lines, MDA-MB-231 and MDA-MB-468 with either Flavopiridol or PD0332991 in combination with overexpression of ER (). Flavopiridol and PD0332991 alone caused increased MS formation in both ER−ve cell lines; however, with the addition of ER expression, a significant decrease was now observed. These data suggest that through the simple re-expression of ER, ER-negative cells now respond to cyclin D1 and CDK4/6 modulation as an ER+ve cell line.
Discussion
We have identified novel roles for cell cycle regulators, cyclin D1 and CDK4/6 in the regulation of migration, mammosphere formation, and ALDH expression, which are indicators of stem-like cell activity. Using 4 cell lines and primary cells from 6 breast cancer patients, we inhibited cyclin D1 and CDK4/6 using both siRNA and inhibitors currently in use in clinical trials. An increase in migration and stem-like cell activity in ER−ve cells, while a decrease in ER+ve cells, was observed. Conversely overexpression of cyclin D1 caused a contrasting effect. Re-expression of ER within ER−ve cell lines was sufficient to overcome the increased migration and mammosphere formation when inhibiting cyclin D1 and the decrease when overexpressing cyclin D1.
Currently within the field of cancer research, much effort is made to inhibit cell proliferation or perhaps to target components within the cell cycle, with the short-term aim of reducing tumor growth and long-term aim of increasing patient survival. Despite many advances in breast cancer treatments, a significant proportion of patients do not respond to current treatment regimens or eventually show recurrence. In a recent study from McClendon and colleagues, the long-term effect of combining PD0332991 and doxorubicin therapy in triple-negative breast cancer cells was examined. The study revealed that although an initial cooperative cytotoxic effect caused by the combination of the 2 drugs was observed, RB-proficient cells were ultimately protected from doxorubicin-mediated cytotoxicity, possibly allowing for a late-onset tumor recurrence.Citation23 In order to improve the outcome of patients, a better understanding of the basic biology of breast cancer is required and an understanding of how cellular characteristics key in the development and progression of breast cancer are interlinked. Key characteristics include the aforementioned proliferation, migration, and stem-like cell activity.
Our data provides a link between proliferation, migration, and stem-like cell activity and has identified cyclin D1 and CDK4/6 as key proteins that coordinate these cellular processes in breast cancer. We have shown that knockdown of cyclin D1 in ER−ve cells increases migration, which is supportive of our previous publication,Citation33 and also increases stem-like cell activity. Stem-like cell activity was primarily assessed by mammosphere formation; however, to give further validation to the effect on stem-like cell activity, ALDH activity, which is a validated marker of breast cancer stem-like cells,Citation29 was also examined. In ER+ve patients, the converse was true, when inhibiting cyclin D1, cells responded with a decrease in migration and stem-like cell activity. CDK4/6 are key proteins that function together by binding to cyclin D1 during the cell cycle, and it was therefore necessary to inhibit both proteins simultaneously. The vast majority of breast cancer samples responded to CDK4/6 inhibition in a similar fashion to cyclin D1, with increased migration and mammosphere formation in ER−ve samples with opposing effects observed in ER+ve samples.
ER expression was highly predictive of breast cancer cell response to cyclin D1 modulation with all samples tested, both cell lines and primary cells displaying an identical response. To confirm the importance of ER within this system, ER was re-expressed in ER−ve breast cancer cell lines in combination with either siRNA to target cyclin D1 or overexpression of cyclin D1. The simple re-introduction of ER into these cell lines reversed the cellular response to cyclin D1 modulation (). This suggests that the balance between proliferation, migration, and stem-like cell activity is tightly regulated, and of most importance, disruption of this balance may have adverse effects. Consideration should therefore be taken when selecting drugs for use in the clinic, as it is essential to understand the mechanism of action of these drugs and the effects within specific subgroups of breast cancer. It has been reported that specific overexpression of cyclin D1 in the mouse mammary gland results in enrichment of genes ranking highly with a chromosomal instability (CIN) signature. This is likely a direct consequence of cyclin D1 expression. Expression of genes promoting CIN, as well as high expression of cyclin D1, were further shown to be enriched in luminal B breast cancers, suggesting cell cycle-targeting drugs to be a promising approach in this particular ER+ breast cancer subgroup.Citation11
To further validate our findings and to highlight the clinical relevance, we used inhibitors of both cyclin D1 and CDK4/6 (Flavopiridol/PD0332991), which are currently undergoing clinical trials in cancer. In 2011, Schwartz et al. performed a phase 1 clinical trial to investigate PD0332991 treatment in 33 patients with retinoblastoma protein-positive advanced solid tumors or non-Hodgkin’s lymphoma. Oral administration was well tolerated, and recommendation for phase 2 testing was made, which is currently underway in a number of cancer types, including metastatic breast cancer.Citation34 Phase 1 clinical trials were performed combining Flavopiridol and Docetaxel treatment in 27 patients with solid tumors including breast cancer. The treatment was tolerated, and the clinical activity was encouraging.Citation35
Using these clinical inhibitors within our study mimicked the observations when using siRNA inhibition. In ER−ve cells, treatment with the inhibitors increased mammosphere formation, while in the ER+ve cells mammosphere formation was decreased. As with the siRNA, re-expression of ER was able to reverse these effects in ER−ve cells. These data suggest that clinical inhibitors designed to inhibit cell cycle proteins have the potential to cause adverse effects in subgroups of patients, specifically ER−ve patients, due to an increase in migratory capacity and stem-like cell population, which, in turn, could increase metastasis and recurrence. In support of our observation, Finn et al. published data investigating the effects of PD0332991 on the proliferation of breast cancer cell lines, identifying both sensitive (MCF-7) and resistant cell lines (MDA-MB-468).Citation20 A further study published by Fry et al. showed that the resistant MDA-MB-468 cell line when grown in a mouse model system showed resistance to PD0332991 treatment with no reduction in tumor growth. A small increase in tumor growth rate compared with control treated mice was observed. In comparison, mice injected with sensitive ZR75.1 cells showed a significant reduction in tumor size.Citation22 Our data suggests that the mechanism by which PD0332991 works is to reduce stem-like cell activity and tumor growth, and if used in patients may reduce metastasis by hampering cells’ ability to migrate and leave the primary tumor site. This treatment should be limited to ER+ve patients, and if used in resistant patients, adverse effects may occur. Following treatment, ER−ve patients may have increased stem-like cell activity, which has been linked to poor prognosis and resistance to radio- and chemotherapy.Citation32,Citation36 Furthermore cells may also be more migratory and therefore have increased capability of metastasis. Breast cancer patients with ER−ve tumors, in particular triple-negative (ER/PR/HER2) tumors, are a subgroup of patients that demonstrate a particularly poor prognosis and currently have limited treatment possibility. Within our experiments, we have shown that treatment with PD0332991 or Flavopiridol is ineffective. However, in combination with re-expression of ER within resistant ER−ve cells is sufficient to overcome the adverse effects of cell cycle inhibition on mammosphere formation and migration. In the presence of ER, ER-negative breast cancer cells are able to respond in a positive manner to cell cycle inhibition with a decrease in migration and stem-like cell activity. For translation to a clinical application, one would treat ER−ve patients with combination therapies to inhibit cell cycle components such as cyclin D1 or CDK4/6 in conjunction with treatments perhaps to induce ER expression. In ER-negative patients, gene methylation has been described which silences protein expression.Citation37 A number of studies have investigated the potential of re-expression ER in ER-negative patients.Citation38-Citation40 In 2010, Li et al. demonstrated that dietary green tea polyphenol (epigallocatechin-3-gallate) was able to reactivate ER expression and worked synergistically with histone deacetylase (HDAC) inhibitors to restore tamoxifen sensitivity.Citation41
The data presented has identified novel roles for cell cycle proteins cyclin D1 and CDK4/6 in breast cancer through the co-coordination of cell cycle, migration, and stem-like cell activity. Various oncogenic pathways, such as, for example Ras, c-Myc, and Neu (ErbB2), have been described to converge on cyclin D1/CDK4 expression, and our results are well in line with the growing body of literature addressing diverse roles of cell cycle proteins.Citation11,Citation13,Citation42,Citation43 Our data also shows contrasting effects of inhibition of cyclin D1 and CDK4/6 dependent upon ER expression in breast cancer cells. Inhibitors of both cyclin D1 and CDK4/6 have been used to treat cancer with some success in the clinic and have more recently been applied to treatment of breast cancer. Our data suggests that in subgroups of breast cancer patients, these drugs may be highly effective, decreasing the potential for metastasis and recurrence by inhibiting both migration and stem-like cell activity. Conversely, these drugs may be ineffective or potentially have adverse effects within ER−ve subgroups of breast cancer. The data does, however, show the potential for the use of the drugs in combination therapies. The simple re-expression or ER conferred cells susceptible to clinical inhibition of cyclin D1 and CDK4/6. Further studies will be needed to investigate the potential of combination drug treatments to combat breast cancer in those patients currently resistant to therapy.
Materials and Methods
Patient samples
Samples were collected with informed consent from 6 breast cancer patients, including both primary and metastatic cells (ER−ve n = 3, ER+ve n = 3). Primary solid tumor samples (obtained through the MCRC Biobank, project ID: 09_GOLA_02) were dissected into 1 mm pieces and incubated at 37 °C for 16 h in 1× Collagenase/Hyaluronidase mixture (STEMCELL Technologies Inc, #07912) in DMEM:F12/15mM HEPES (Sigma-Aldrich Co, #D6421). Pleural effusion and ascites samples were collected with informed consent from patients with metastatic breast cancer during standard drain protocol (Ethics # 05/Q1403/159). Following extraction from tissue (as detailed above) or collection of metastatic fluid, cells were collected by centrifugation at 800 g and resuspended in PBS. Blood cells were removed by centrifugation through 0.5 volumes of Lymphoprep solution (Axis-Shield plc, #1114544) at 600 g. Cells were cultured in DMEM:F12/20% FCS/0.1% non-essential amino acid solution/2.5 mM L-glutamine/PenStrep (P/S).
Cell lines
All cell lines were purchased from ATCC; MDA-MB-231, MDA-MB-468 (ER-negative) MCF7, and T47D (ER-positive). Cell lines were authenticated by multiplex PCR assay using the AmpFlSTR Indentifiler PCR amplification kit (Applied Biosystems, Life Technologies Corporation, #4322288) and confirmed as mycoplasma-free. MDA-MB-231 and MDA-MB-468 were cultured in RPMI complete medium (RPMI/10% FCS/1% Sodium pyruvate/2 mM L-glutamine/PenStrep) while MCF7 and T47D were grown in DMEM complete medium (DMEM/10% FCS/2 mM L-glutamine/PenStrep). Cells were maintained in a humidified incubator at 37 °C at an atmospheric pressure of 5% (v/v) carbon dioxide/air.
siRNA and vector transfections
Four × 105 cells were seeded in a 28.3 cm2 culture dish with either DMEM or RPMI for 24 h. The media was subsequently removed and P/S-free SFM (serum free media) added along with 1 ml of siRNA solution (Opti-MEM®, Gibco, #11058021, Lipofectamine® 2000, Invitrogen Life Technologies, #11668-019) giving a final concentration of 40 nmol/L oligonucleotides. Six hours after transfection, SFM was replaced with SM (serum media) and cells were allowed to grow for 24 h before harvesting for subsequent analysis. ONTARGETplus SMARTpool siRNA reagents targeting cyclin D1 (Dharmacon RNA Technologies, #LQ003210 excluding duplex #J-003210-15 due to non-specific effects), CDK4 (#L-003238) in combination with CDK6 (#L-003240) and a non-targeting pool was used as negative control (#D-001810-10).
For cyclin D1 vector experiments, 3 μg of GFP-tagged vector with an insert of the ORF of cyclin D1, or 3 μg GFP only (control) was used (OriGene Technologies Inc, #RG204957). For ER-α transient transfections, cells were transfected with either 3 μg pVP16-ER vector (Addgene, #11351) or pVP16 only (control) (donation from Keith Brennan). All transfections were using lipofectamine according to manufacturer’s protocol.
Inhibitors of cyclin D1 and CDK4/6 in use in clinical trials
Breast cancer cells were treated with a pan-CDK inhibitor also inhibiting cyclin D1 (Flavopiridol, Selleckchem, #S2679)Citation44 and a CDK4/6 inhibitor (PD0332991, Selleckchem, #S1116)Citation22 that are currently undergoing clinical trials in breast cancer. Inhibitors were added to mammosphere (MS) media for the subsequent culture of mammospheres. Inhibitors were used at a range of concentrations. In cell lines, Flavopiridol was used at 20 ng/ml and 50 ng/ml, while in primary human cells it was used at concentrations of 2 ng/ml and 10 ng/ml. PD0332991 was used at concentrations of 0 ng/ml, 2 ng/ml, and 10 ng/ml in both cell lines and primary human cells.
Mammosphere culture
A single cell suspension was prepared using enzymatic (1× Trypsin-EDTA, Sigma Aldrich, #T3924) and manual disaggregation (25-gauge needle). Cells were plated at a density of 500 cells/cm2 in non-adherent conditions in mammosphere medium (DMEM-F12/B27/20 ng/ml EGF/PenStrep). Cells were grown for 5 d and maintained in a humidified incubator at 37 °C at an atmospheric pressure in 5% (v/v) carbon dioxide/air. Spheres >50 μm were counted using an eye piece graticule.
Migration assay
Transwell chambers with a diameter of 6.5 mm and a pore size of 8 µm were used to assess migration (Corning, Inc, #3422). Migration chambers were incubated with 150 μl serum-free media for an initial equilibrium period of 1 h. Cells (transfected the day before) were resuspended in serum-free media, and 50 000 cells were added to each migration chamber. Next, chambers were placed into wells containing 600 μl 10% FCS medium, and cells were allowed to migrate for 5 h (MDA-MB-231) or overnight (MCF7, T47D, MDA-MB-468, and primary breast cells). Cells that had not migrated were removed with a cotton swab, whereas the migrated cells situated on the lower side of membranes were fixed in 4% paraformaldehyde for 25 min. Membranes with the migrated cells were mounted on glass slides for DAPI staining and counted using a fluorescent microscope (cells in six ×10 magnification fields were counted). Experiments were repeated at least three times. The percentage of cells that had migrated was then calculated.
Western blotting
Protein was loaded onto an SDS–polyacrylamide gel and transferred to Hybond-C Extra nitrocellulose membrane (Amersham, GE Healthcare, Life Sciences, #RPN303E). Primary antibodies included: cyclin D1 (Dako, # M7155), CDK4 (Chemicon International Inc, #MAB8879), CDK6 (Cell Signaling Technology Inc, #3136), SP1-ERα (Thermo Fisher Scientific Inc, #RM-9101-SO), actin (Santa Cruz Biotechnology, #sc-1616).
Statistical methods
Throughout the paper data are represented as mean ± SEM taken over a minimum of 3 independent experiments. Statistical significance was measured using parametric testing, assuming equal variance, with standard t tests for 2 paired samples used to assess difference between test and control samples.
| Abbreviations: | ||
| ER | = | estrogen receptor |
| CDK | = | cyclin-dependent kinase |
| ALDH | = | aldehyde dehydrogenase |
| MS | = | mammosphere |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55:74 - 108; http://dx.doi.org/10.3322/canjclin.55.2.74; PMID: 15761078
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 365:1687 - 717; http://dx.doi.org/10.1016/S0140-6736(05)66544-0; PMID: 15894097
- Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. Clin Cancer Res 2003; 9:1980 - 9; PMID: 12796359
- Blanco G, Holli K, Heikkinen M, Kallioniemi OP, Taskinen P. Prognostic factors in recurrent breast cancer: relationships to site of recurrence, disease-free interval, female sex steroid receptors, ploidy and histological malignancy grading. Br J Cancer 1990; 62:142 - 6; http://dx.doi.org/10.1038/bjc.1990.247; PMID: 2390476
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2:563 - 72; http://dx.doi.org/10.1038/nrc865; PMID: 12154349
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999; 98:859 - 69; http://dx.doi.org/10.1016/S0092-8674(00)81519-6; PMID: 10499802
- Courjal F, Louason G, Speiser P, Katsaros D, Zeillinger R, Theillet C. Cyclin gene amplification and overexpression in breast and ovarian cancers: evidence for the selection of cyclin D1 in breast and cyclin E in ovarian tumors. Int J Cancer 1996; 69:247 - 53; http://dx.doi.org/10.1002/(SICI)1097-0215(19960822)69:4<247::AID-IJC1>3.0.CO;2-X; PMID: 8797862
- Gillett C, Smith P, Gregory W, Richards M, Millis R, Peters G, et al. Cyclin D1 and prognosis in human breast cancer. Int J Cancer 1996; 69:92 - 9; http://dx.doi.org/10.1002/(SICI)1097-0215(19960422)69:2<92::AID-IJC4>3.0.CO;2-Q; PMID: 8608989
- McIntosh GG, Anderson JJ, Milton I, Steward M, Parr AH, Thomas MD, et al. Determination of the prognostic value of cyclin D1 overexpression in breast cancer. Oncogene 1995; 11:885 - 91; PMID: 7675447
- Casimiro MC, Pestell RG. Cyclin d1 induces chromosomal instability. Oncotarget 2012; 3:224 - 5; PMID: 22538871
- Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle 2012; 11:4642 - 9; http://dx.doi.org/10.4161/cc.22937; PMID: 23187803
- Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci USA 2006; 103:11567 - 72; http://dx.doi.org/10.1073/pnas.0603363103; PMID: 16864783
- Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol 2001; 21:4773 - 84; http://dx.doi.org/10.1128/MCB.21.14.4773-4784.2001; PMID: 11416152
- Ezhevsky SA, Nagahara H, Vocero-Akbani AM, Gius DR, Wei MC, Dowdy SF. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl Acad Sci USA 1997; 94:10699 - 704; http://dx.doi.org/10.1073/pnas.94.20.10699; PMID: 9380698
- Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 1998; 18:753 - 61; PMID: 9447971
- Reddy HK, Mettus RV, Rane SG, Graña X, Litvin J, Reddy EP. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res 2005; 65:10174 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-2639; PMID: 16288002
- Lapointe J, Lachance Y, Labrie Y, Labrie C. A p18 mutant defective in CDK6 binding in human breast cancer cells. Cancer Res 1996; 56:4586 - 9; PMID: 8840966
- Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012; 11:2756 - 61; http://dx.doi.org/10.4161/cc.21195; PMID: 22767154
- Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009; 11:R77; http://dx.doi.org/10.1186/bcr2419; PMID: 19874578
- Fry DW, Bedford DC, Harvey PH, Fritsch A, Keller PR, Wu Z, et al. Cell cycle and biochemical effects of PD 0183812. A potent inhibitor of the cyclin D-dependent kinases CDK4 and CDK6. J Biol Chem 2001; 276:16617 - 23; http://dx.doi.org/10.1074/jbc.M008867200; PMID: 11278443
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3:1427 - 38; PMID: 15542782
- McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle 2012; 11:2747 - 55; http://dx.doi.org/10.4161/cc.21127; PMID: 22751436
- Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res 2010; 70:5649 - 69; http://dx.doi.org/10.1158/0008-5472.CAN-10-1040; PMID: 20610625
- Cheng CW, Wu PE, Yu JC, Huang CS, Yue CT, Wu CW, et al. Mechanisms of inactivation of E-cadherin in breast carcinoma: modification of the two-hit hypothesis of tumor suppressor gene. Oncogene 2001; 20:3814 - 23; http://dx.doi.org/10.1038/sj.onc.1204505; PMID: 11439345
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117:927 - 39; http://dx.doi.org/10.1016/j.cell.2004.06.006; PMID: 15210113
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983 - 8; http://dx.doi.org/10.1073/pnas.0530291100; PMID: 12629218
- Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res 2009; 69:1302 - 13; http://dx.doi.org/10.1158/0008-5472.CAN-08-2741; PMID: 19190339
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007; 1:555 - 67; http://dx.doi.org/10.1016/j.stem.2007.08.014; PMID: 18371393
- Lai VK, Ashraf M, Jiang S, Haider K. MicroRNA-143 is a critical regulator of cell cycle activity in stem cells with co-overexpression of Akt and angiopoietin-1 via transcriptional regulation of Erk5/cyclin D1 signaling. Cell Cycle 2012; 11:767 - 77; http://dx.doi.org/10.4161/cc.11.4.19211; PMID: 22374674
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100:672 - 9; http://dx.doi.org/10.1093/jnci/djn123; PMID: 18445819
- Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 2006; 98:1777 - 85; http://dx.doi.org/10.1093/jnci/djj495; PMID: 17179479
- Lehn S, Tobin NP, Berglund P, Nilsson K, Sims AH, Jirström K, et al. Down-regulation of the oncogene cyclin D1 increases migratory capacity in breast cancer and is linked to unfavorable prognostic features. Am J Pathol 2010; 177:2886 - 97; http://dx.doi.org/10.2353/ajpath.2010.100303; PMID: 20971731
- Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer 2011; 104:1862 - 8; http://dx.doi.org/10.1038/bjc.2011.177; PMID: 21610706
- Fornier MN, Rathkopf D, Shah M, Patil S, O’Reilly E, Tse AN, et al. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res 2007; 13:5841 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-07-1218; PMID: 17908977
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007; 131:1109 - 23; http://dx.doi.org/10.1016/j.cell.2007.10.054; PMID: 18083101
- Lapidus RG, Ferguson AT, Ottaviano YL, Parl FF, Smith HS, Weitzman SA, et al. Methylation of estrogen and progesterone receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin Cancer Res 1996; 2:805 - 10; PMID: 9816234
- Du J, Zhou N, Liu H, Jiang F, Wang Y, Hu C, et al. Arsenic induces functional re-expression of estrogen receptor α by demethylation of DNA in estrogen receptor-negative human breast cancer. PLoS One 2012; 7:e35957; http://dx.doi.org/10.1371/journal.pone.0035957; PMID: 22558281
- Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast Cancer Res Treat 2003; 81:177 - 86; http://dx.doi.org/10.1023/A:1026146524737; PMID: 14620913
- Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res 2001; 61:7025 - 9; PMID: 11585728
- Li Y, Yuan YY, Meeran SM, Tollefsbol TO. Synergistic epigenetic reactivation of estrogen receptor-α (ERα) by combined green tea polyphenol and histone deacetylase inhibitor in ERα-negative breast cancer cells. Mol Cancer 2010; 9:274; http://dx.doi.org/10.1186/1476-4598-9-274; PMID: 20946668
- Jirawatnotai S, Hu Y, Livingston DM, Sicinski P. Proteomic identification of a direct role for cyclin d1 in DNA damage repair. Cancer Res 2012; 72:4289 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-11-3549; PMID: 22915759
- Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle 2011; 10:57 - 67; http://dx.doi.org/10.4161/cc.10.1.14449; PMID: 21200143
- Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ, Pestell R, et al. Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res 1999; 59:4634 - 41; PMID: 10493518