Abstract
TP53’s role as guardian of the genome diminishes with age, as the probability of mutation increases. Previous studies have shown an association between p53 gene mutations and cancer. However, the role of somatic TP53 mutations in the steep rise in cancer rates with aging has not been investigated at a population level. This relationship was quantified using the International Agency for Research on Cancer (IARC) TP53 and GLOBOCAN cancer databases. The power function exponent of the cancer rate was calculated for 5-y age-standardized incidence or mortality rates for up to 25 cancer sites occurring in adults of median age 42 to 72 y. Linear regression analysis of the mean percentage of a cancer’s TP53 mutations and the corresponding cancer exponent was conducted for four populations: worldwide, Japan, Western Europe, and the United States. Significant associations (P ≤ 0.05) were found for incidence rates but not mortality rates. Regardless of the population studied, positive associations were found for all cancer sites, with more significant associations for solid tumors, excluding the outlier prostate cancer or sex-related tumors. Worldwide and Japanese populations yielded P values as low as 0.002 and 0.005, respectively. For the United States, a significant association was apparent only when analysis utilized the Surveillance, Epidemiology, and End Results (SEER) database. This study found that TP53 mutations accounts for approximately one-quarter and one-third of the aging-related rise in the worldwide and Japanese incidence of all cancers, respectively. These significant associations between TP53 mutations and the rapid rise in cancer incidence with aging, considered with previously published literature, support a causal role for TP53 according to the Bradford-Hill criteria. However, questions remain concerning the contribution of TP53 mutations to neoplastic development and the role of factors such as genetic instability, obesity, and gene deficiencies other than TP53 that reduce p53 activity.
Introduction
The TP53 gene encoding the p53 protein has been called the guardian of the genomeCitation1 because of its role in mediating tumor suppression by irreversible cell cycle arrest (replicative senescence) and apoptosis of cells with damaged DNA. However, TP53 is the most commonly lost gene function in human cancers, with the great majority (>98%) of TP53 gene mutations being acquired somatically.Citation2 These TP53 mutations are chiefly point missense substitutions (75%), with other frequent alterations including frameshift insertions and deletions (9%), nonsense mutations (7%), and silent mutations (5%).Citation3 The effects of these mutations are complex, as in addition to decreased tumor suppression and depressed policing of oncogenic signaling, the loss of TP53 may under some circumstances aid oncogenesis by endowing cancer stem cells with a proliferative (non-quiescent) and immortal gain of function.Citation4-Citation7
The mutational inactivation of TP53 and its effect on the neoplastic process is also complex and depends on the grade at which the TP53 mutation occurs. Mutations early in the process aid the transformation from benign stage to malignant form, leading to the Warburg effect and reliance on glycolysis. Later alterations of TP53 can influence the transition from a non-invasive lesion to an invasive one.Citation8 Two animal studies published in 2007Citation9,Citation10 determined that TP53 mutations promote not only tumor development, but also established tumors. Furthermore, these experiments demonstrated that TP53 restoration reversed the neoplastic process: Ventura et al.Citation9 showed that lymphoma cells underwent apoptosis and sarcomas displayed replicative senescence, while Xue et al.Citation10 found that liver tumor cells cleared rapidly, with apparent activation of the innate immune system. Surprisingly, not all mutations in TP53 are inactivating; for example, abnormal accumulation (overexpression) of p53 is found in a few cancers, e.g., Hodgkin disease and breast cancer.Citation11,Citation12 Furthermore, one study found that p53 can exert pro- or anti- apoptotic effects, depending on cellular context.Citation13
Cancer incidence rates in adults typically rise steeply with age, with the increased incidence starting in the reproductive period and decelerating or declining in old age, about 70 y and over ( based on Canadian data).Citation14 Mortality rates also show an age-dependent trend similar to incidence rates but shifted by a time lag between the age at clinical manifestation and death from the disease (). For many types of cancer, the incidence and mortality age-standardized rate (ASR; typically a weighted mean of age-specific rates for 100 000 of a standard population) among adults (excluding the very old) can be represented by a power function:
Figure 1. Annual incidence and mortality ASR of all cancers. (A) Incidence in 2007 (I) and average mortality in 2004–2008 (M) rates per 100 000 population (Statistics Canada)Citation14 for males, females, and both sexes (mixed). (B) Log-log plot of cancer incidence ASR and age for both sexes combined, showing the cancer exponent trend line.
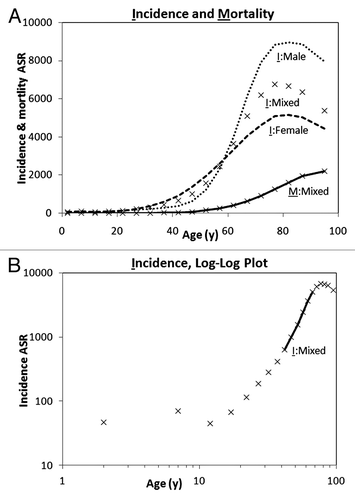
ASR(t) = c tK (1)
where t is the age in years, K is the “cancer exponent,” and c is a constant. A plot of log(ASR) vs. log(t) is a straight line with a slope of K ():
log(ASR) = log(c) + K log(t) (2)
Armitage and DollCitation15 hypothesized that a discrete number of mutational stages could be estimated from a rounded value of the cancer exponent plus 1. SRT such as those of the prostate, ovary, and uterus exhibit variable hormone and metabolism dependence and can depart from the power function. Breast tumors, for example, are initially dependent on estrogens or obesity-related metabolism for growth and progression; their incidence can be much reduced after menopause in populations that have not adopted a western-like lifestyle.Citation16,Citation17
TP53 is the subject of nearly 300 000 publications,Citation18 yet a quantitative estimate of the contribution of TP53 mutations to cancer incidence and mortality is lacking. This study evaluates the role of somatic TP53 mutations on the steep aging-related rise in regional cancer rates employing large, international, population-based databases. It first provides a quantitative analysis of the rise of adult cancer rates with age to determine whether there are significant associations with somatic TP53 mutations in those cancers and, second, considers whether these associations, in combination with the evidence of published studies, support a causal role.
Results
Prevalence of TP53 mutations in cancers
On the basis of IARC GLOBOCAN and TP53 databases, TP53 mutations are present in an estimated 29% of all cancer cases diagnosed globally (n = 25, ~92% of total incidence ASR), and 32% of solid tumor cases, i.e., excluding leukemia and multiple myeloma (n = 23, ~88% of total incidence ASR). When hormone-associated SRT (~27% of total incidence ASR) are excluded, a similar TP53 mutation rate of 29%, and slightly greater rate of 34%, is found in all cancer cases and solid tumor cases diagnosed, respectively. Therefore, the mutations are found in about 1 in 3 of all malignant tumors occurring in the world population.
Regional relationship between TP53 mutations and cancer incidence and mortality
Worldwide
For global data, the 27 cancer exponent (slope of log ASR vs. log age plot) values varied from -1.7 (testicular cancer) to 11.2 (prostate cancer) for incidence ASR and from 1.5 (cervix uteri cancer) to 11.0 (prostate cancer) for mortality ASR (). These 27 cancer exponents were obtained from linear regression plots whose mean R value was 0.97 ± 0.07 SD. For only 4 cancer sites (Kaposi sarcoma, nasopharynx, cervix uteri, and testis cancer) was the relationship weaker than R < 0.95 ().
Table 1. Analysis of worldwide cancer exponent values for IARC GLOBOCAN incidence age-standardized rates of cancer (6 five-year age groups; both sexes except for SRT) and IARC TP53 mutation rates, for various types of cancer by topographic or morphologic sites
For the mortality ASR, for all cancers and for solid tumors (both sexes or male and female separately), including and excluding SRT, regression analysis yielded no significant associations (P > 0.05) between the percentage of samples with TP53 mutations and cancer exponents.
The same regression analyses were conducted separately for incidence ASR for males and females. Prostate cancer (~8% of total incidence ASR) was an outlier (standardized residual ≥3.0), with its highest cancer exponent (11.2) nearly double the next highest. This extremely high exponent may be related to benign nodular prostatic hyperplasia being found in almost all men after 50 y.19 A significant association (R2 = 20%, P = 0.03) was found for females, which included 4 SRT, yet non-significant for males (R2 = 6%, P = 0.3), unless prostate cancer was excluded (R2 = 23%, P = 0.03; ). Hence, the regression model indicates by R2 that 20% or more of the cancer exponent variability is explained by TP53 mutations.
Table 2. Regression parameters for cancer exponents from incidence ASR for all cancers and solid tumors and TP53 mutation rates for individuals aged 40–44 y to 65–69 y, analyses conducted worldwide and in three geographical regions (IARC GLOBOCAN data with addition of SEER data for the United States)
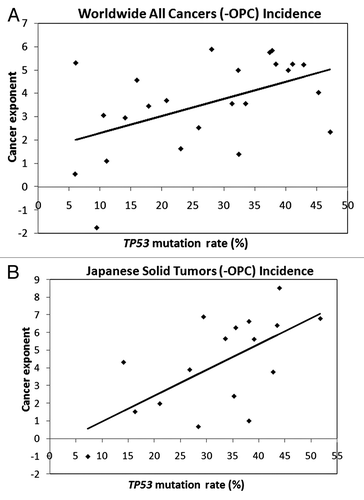
For all cancers or solid tumors only, of both sexes combined, associations between TP53 mutation rates and cancer exponents were non-significant if OPC was included yet significant if OPC was excluded (R2 = 24%, P = 0.01, all cancers, and R2 = 38%, P = 0.002, solid tumors). Similarly, TP53 mutation rates and cancer exponents were non-significant if SRT were included, yet significant for solid tumors only if SRT were excluded (R2 = 17%, P = 0.08, all cancers and R2 = 31%, P = 0.02, solid tumors).
Figure 2. Linear regression trend lines for IARC incidence ASR and IARC TP53 mutation rates for individuals aged 40–44 y to 65–69 y. (A) the worldwide incidence of 24 all cancers excluding OPC, R = 0.49, CI = 0.11–0.75, P = 0.01. (B) The Japanese incidence of 17 solid tumors, excluding OPC, R = 0.64, CI = 0.24–0.86, P = 0.005.
Major cancer types originate as epithelial cells (carcinomas), hematopoietic cells (leukemia, lymphoma, myeloma), germ cells (testicular cancers), and mesenchymal cells (sarcomas). I compared TP53 mutations in 20 primarily epithelial tumor sites and 4 non-epithelial tumor sites (leukemia, multiple myeloma, non-Hodgkin lymphoma, testis), excluding brain cancers, which have exceptional morphologic diversity, and Hodgkin lymphoma, which has less than 50 TP53 samples. A highly statistically significant difference (t test, P = 0.002; P = 0.0002 including Hodgkin lymphoma) was found, with TP53 mutation rates of 30 ± 12% SD in epithelial tumors and 12 ± 6% SD in non-epithelial tumors.
Japan, Western Europe, and United States
Similar to worldwide data, no significant link was identified between TP53 mutations and mortality cancer exponents in each of these three regions. Prostate cancer was an outlier for Japan and Western Europe but not for the United States (standardized residual ~2.6). For all 3 regions, relationships between the TP53 mutation rates and incidence cancer exponents for all cancers and solid tumors were non-significant when OPC or SRT were included.
For Japan, when OPC was excluded there was a stronger and highly significant association for all cancer sites (R2 = 38%, P = 0.006) and solid tumors, (R2 = 41%, P = 0.005). However, when SRT were excluded, there was a weaker yet significant association (R2 = 34%, P = 0.03) for all cancer sites and an association, although not quite significant (R2 = 29%, P = 0.06), for solid tumors.
For Western Europe, the reverse was true for SRT exclusion, with a weakly significant association (R2 = 28%, P = 0.04) for solid tumors and a non-significant association (R2 = 18%, P = 0.09) for all cancers. When OPC was excluded for Western Europe there was a significant association for solid tumor sites only (R2 = 16%, P = 0.07 all cancers, R2 = 24%, P = 0.03 solid tumors).
Similar regression analyses for all cancers and solid cancers utilizing IARC GLOBOCAN United States data showed no evidence of an association (P ≥ 0.2). Prostate and breast cancer had the major standardized residuals (~2.7 and −1.4, respectively). Of the three regions, IARC data for the United States had the smallest number of all cancer and solid tumor sites with an adequate number of TP53 samples (17 and 15 sites, respectively). I therefore employed the SEER cancer data set to increase the number of sites with adequate IARC TP53 sampling data for all cancers and solid tumors (20 and 17 sites, respectively). In this case, a weakly significant association (R2 = 30%, P = 0.05) was found for the log-log plot of the percentage of samples with TP53 mutations and cancer exponents for solid tumors when SRT were excluded.
The linear regression analyses of the TP53 mutation rates and cancer exponents of all cancers and solid tumors, with or without SRT, were compared for paired geographical regions (Japan:Western Europe; Western Europe:United States; and Japan:United States) by ANCOVA. For every regression model, slopes and means did not differ significantly between paired regions. Similarly, after exclusion of prostate cancer, only the slopes for Japan:United States differed at a level that was close to significance or weakly significant (P = 0.04 all cancers, P = 0.07 solid tumors). These results indicate that the rise in cancer incidence with aging is a similar function of the TP53 mutation rate regardless of geographical region, except that TP53 mutations appear to have a greater influence on cancer rates in Japan than the United States.
Relationship between TP53 mutation rates and cancer incidence by country
No significant associations (P ≥ 0.1 in all cases) were found for TP53 mutation rates and their respective cancer exponents for 5 major topographic cancer sites (ni: brain, breast, colorectum, liver, and lung) in different countries (). Similarly, no association (P ≥ 0.2 in all sites) was found between the TP53 mutations and the cancer exponent for the principal morphologic tumors of the 5 sites.
Table 3. Regression analysis by country of cancer exponents from incidence ASR for 5 major topographic cancer sites (and prostate cancer) and their corresponding principal morphologic sites, vs. TP53 mutation rates or body mass index (BMI) for individuals aged 40–44 y to 65–69 y (both sexes, except for breast and prostate)
The variation in the international TP53 mutation rates for brain, breast, colorectum, liver, and lung is small, with SD values of 31, 22, 20, 51, and 40% of the mean, respectively: liver has the largest percentage TP53 mutation rate variation (mean 24 ± 12 SD) between countries. Except for the breast cancer exponent, the respective cancer exponents generally have similar or even smaller variation than the mutation rates, with SD values for brain, breast, colorectum, liver, and lung of 18, 55, 10, 24, and 16% of the mean, respectively. The variation of TP53 and cancer exponents among countries of the 5 principal morphologic tumors is of the same order as that of the corresponding topographic sites.
Lastly, prostate cancer, with only 5 countries having ≥50 TP53 samples, yielded a non-significant association (R2 = 2%, P = 0.8) between percentage TP53 mutation rates, mean 13 ± 10 SD, and cancer exponents, mean 11.7 ± 1.7 SD.
Relationship between body mass index and cancer incidence by country
A non-TP53 influence was explored for brain, breast, colorectum, liver, and lung topographic cancer sites and their principal morphologic tumors. Only linear regression analyses of breast cancer and breast carcinoma yielded significant associations (R2 = 42%, P = 0.001, cancer and R2 = 52%, P = 0.003, carcinoma) between age-standardized body mass indexCitation20 and cancer exponents in 22 and 15 countries, respectively (). Limited to 5 countries with adequate TP53 data, or extended to 24 countries (see “Materials and Methods”), prostate cancer, unlike breast cancer, gave a non-significant association with body mass index (n = 5, R2 = 23%, P = 0.4; n = 24, R2 = <0.1%, P = 0.9).
Discussion
Databases that aggregate data from national or international sources, such as the IARC and SEER databases used in this study, have intrinsic limitations due to different resources, methods of interpretation, and analysis.Citation21 Notwithstanding these limitations and the analytical methods employed in this study, the linear regression analyses led to quantification of significant relationships between TP53 mutation rates and the incidence cancer exponent.
To assess a potential causal role for TP53 mutations in the aging-related rise in cancer rates, I have used the findings of this study and previous studies to evaluate if the 9 criteria for causation established by epidemiologist Bradford-Hill are satisfiedCitation22,Citation23 (). The significant associations found address the “strength of association” criterion. In addition, the “consistency” Bradford-Hill criterion is met by the fact that the slope and intercept values of the TP53 mutation rates and the cancer exponents are unfailingly positive and of the same magnitude regardless of the geographical area or the cancer categories analyzed in this study. While large uncertainties remain, this study shows that worldwide (), about a quarter of the aging-related exponential rise in the diagnosed tumors at all sites, excluding OPC, could be assigned to TP53 mutations. TP53 mutations have a smaller involvement in Western Europe and the United States, and a larger involvement in Japan (accounting for approximately one-third of the aging-related rise), perhaps because of other factors such as the low rate of obesity.
Table 4.TP53 mutations and the aging-related rise in cancer incidence: Bradford-Hill criteria assessmentCitation22
Unlike the 3 other regions analyzed solely on the basis of IARC databases, the United States data for TP53 mutation rates and cancer exponents did not yield a significant relationship either for incidence of all cancer sites or solid tumors, even when SRT were excluded (). Of the 4 regions, United States data in the IARC TP53 database included the lowest number of IARC GLOBOCAN cancer sites, adequately represented by 50 or more TP53 mutation samples (all cancer sites, n = 17). However, I obtained a weakly significant association for solid cancers excluding SRT by alternatively employing SEER rather than IARC cancer data for the United States, increasing the number of all cancer sites to 20, supporting the importance of including a high number of cancer sites in this type of analysis.
Despite the associations found between the incidence cancer exponents K of various cancer sites n and the TP53 mutation rates in specific geographical regions ri , i.e., TP53(n)ri vs. K(n) ri, similar significant relationships were absent between the TP53 mutation rates in various countries N and the cancer exponents of specific cancer sites ni , i.e., K(N)ni vs. TP53(N)ni , or their principal tumors, i.e., TP53(N)Ti vs. K(N)Ti. One possible reason for this discrepancy is the larger range of TP53 mutation rates among cancer sites globally, varying ~7-fold from a low value of 6% for cervix uteri to a high value of 47% for ovarian cancers (). However, the range is narrower for specific cancer sites; for example, the TP53 mutation rate for breast cancer varies between 7% for Greece and 29% for Italy.
A second possible reason for the lack of association is that additional factors, other than differing TP53 mutations by country, may have a strong influence on their cancer exponents. For example, for breast cancer, the cancer exponents of 5 Asian countries (China, Chinese Taiwan, India, Japan, and Korea) were statistically much smaller (~10-fold) than the values from the 17 non-Asian countries (means 0.2 ± 0.8 SD, 2.4 ± 0.4 SD, respectively; t test, P = 0.003). However, the corresponding TP53 mutation rates are similar for these Asian and non-Asian countries (means 23 ± 7% SD, 23 ± 5% SD, respectively; t test, P = 0.999). An exploratory analysis of the relation between the body mass index and breast cancer exponents in various countries yielded a highly significant association, raising the possibility that obesity has a greater influence than TP53 mutations for breast and perhaps other cancers.Citation16,Citation17,Citation38
There are good grounds for Bradford-Hill’s “specificity” criterion (), as cancer susceptibility is an inherited property of Li-Fraumeni syndrome (characterized by germ line mutation of one TP53 allele),Citation24 in which the age of onset of cancer is directly related to the TP53 transactivation functionality.Citation28 Conversely, above-normal levels of p53 likely play a role in reducing cancer incidence but also lifespan in those affected by Huntington disease.Citation39
A stronger relationship between all cancer TP53 mutation rates and cancer exponents occurs when excluding prostate cancer or solid cancers, the latter mainly due to the removal of multiple myeloma. These two cancers commonly have analogous factors to TP53 mutations that reduce p53 protein activity. In the case of prostate cancer, up to 70% of tumors are devoid of a PTEN allele that regulates p53 stability.Citation40 In addition to the TP53 mutational status of multiple myeloma being controversial, its TP53/Mdm2 signaling pathways can be impaired by epigenetic silencing of p53-inducible microRNAs.Citation41
No significant associations between TP53 mutation rates and mortality cancer exponents were found. Yet previous studies have found an effect of TP53 mutations on both cancer prognosis and outcomes, influencing not only tumor initiation, but also metabolism, angiogenesis, immunology, and invasion.Citation28,Citation29,Citation42 In , I note studies that support the Bradford-Hill criterion of “temporality”, in which TP53 mutations precede cancer incidence.Citation25,Citation26 However, other studies, for example involving adenoma, a colonic polyp that is a precursor of colorectal cancer, indicate ambiguity as to when in tumor development a mutation in the TP53 gene is acquired and the tumor transforms into an invasive carcinoma.Citation43 For acute lymphoblastic leukemia patients, TP53 mutations are about 10-fold more common at relapse than at initial presentation, possibly because TP53 mutations occur during the course of treatment.Citation44 Nevertheless, in acute myeloid leukemia patients, a low TP53 mutation rate is found both at presentation and relapse. Therefore, in this leukemia TP53 mutations more likely occur before diagnosis.
Many factors have been proffered to explain the steep increase in the incidence of cancer with aging. Possible factors not specifically related to TP53 include obesity, viruses, cumulative exposure to carcinogens such as radiation, immunological decline, less effective DNA repair; exhaustion of stem cells, and accumulation of senescent cells, pre-existing lesions, and somatic mutations.Citation45,Citation46 Factors that can be related to p53-gene mutations include imbalance between cell birth and death, chromosomal instability and gene amplification.Citation47 In addition, Feng et al.Citation48 have identified a TP53 factor in which mutation changes may only partially represent the whole TP53 effect with aging. They found the TP53 transcriptional activity and p53-dependent apoptosis response become less efficient in old age by exposing mice to gamma-radiation (5 Gy). The declining efficiency of p53-mediated apoptosis, senescence, or repair in retort to stress is one plausible explanation among others behind the general rise in chromosomal defects, somatic DNA mutations, and tumor incidence in older individuals.Citation45 A cascade of mutations may arise not only from the functional loss of a gatekeeper gene, such as TP53 and other TP53 pathway mutations (BCL2, MDM2, MYC, and RAS),Citation35-Citation37 but by alternative means, such as telomere loss, defective DNA repair (i.e., caretaker gene mutations), and DNA-damaging agents such as radiation or viruses. Therefore, the high prevalence of the TP53 mutations among the multiple mutations of cancers, together with the almost universal occurrence of telomerase activation, implies that TP53 mutations may be linked to the neoplastic process, which preferentially utilizes glycolytic pathwaysCitation29 and bypasses TP53-induced senescenceCitation4 to achieve immortality.
Somatic mutations in different tumor types can range from a few to more than a thousand,Citation49,Citation50 although most are harmless passenger mutations, not driver mutations and therefore not cancer genes.Citation51 The number of driver mutations in general (rather than TP53 in particular) may be associated with the cancer exponent of incident cancer rates. For example, testicular cancer and common leukemias (e.g., acute lymphoblastic leukemia and acute myeloid leukemia) are associated with few somatic mutations,Citation49 infrequent presence of TP53 mutations (≤11%),Citation44 and cancer exponents with a relatively low value (). Conversely, epithelial cancer sites, which are dominant in adult humans, have on average a 2.5-fold higher rate of TP53 mutations compared with non-epithelial cancer sites, providing a possible explanation for the preferential promotion of epithelial cancers.
Genomic instability is particularly effective at advancing the multistage process of inducing epithelial cancers, as shown in humans and animal studies.Citation47,Citation52 Genomic instability is also a hallmark of cancer cells generally, manifested by chromosomal mutations and aneuploidy, the latter being the most common chromosomal defect associated with cancer.Citation30-Citation32 An important question is whether TP53 mutations are an important cause or result of genetic instability.Citation27,Citation53 Indirect support for a causal role arises from the fact that childhood cancers have a lower rate of TP53 mutations, fewer somatic mutations, and less evidence of genomic instability than adult cancers.Citation54,Citation55 An experiment in mice by Schmitt et al.Citation32 supports a causal role of TP53 more directly, finding that TP53 loss sets off a chain reaction of genetic instability and aneuploidy, leading to lymphoma.
Hence, this assessment of the association between the candidate causative agent, TP53 mutations, and the aging-related rise in cancer finds grounds for a causal role based on my results and previously reported literature, meeting all 9 Bradford-Hill criteria ().Citation22 This assessment was performed in a manner similar to Gazdar et al.,Citation56 who showed that 8 of the 9 Bradford-Hill criteria were addressed for the universally accepted relationship between the human papillomavirus and cervical cancer. Of these 8 criteria, the “strength of association” for this virus is very persuasive, present in more than 90% of cervical tumors. This is higher than the percentage of TP53 mutations found in cancers in general, yet similar to TP53 mutations in particular tumors, e.g., 70% in small cell lung cancers.Citation21 For human papillomavirus and cervical cancer, the “biological gradient” is unclear, whereas significant associations have been recorded () for the presence of TP53 mutations and the rise in adult cancer incidence. Although TP53 mutations and consequent TP53 inactivation satisfy all 9 Bradford-Hill criteria for causality, there is still reasonable doubt about TP53’s role in the aging-related rise in cancer. Other events, such as genetic instability and unidentified aspects of the aging process, may be complicit or indeed more important than TP53.
In summary, numerous publications have linked TP53 mutational inactivation with cancer, yet an overall quantification of the influence of this genetic mutation on the general rise in cancer incidence with aging has not previously been reported. In this work, significant relationships were identified between somatic TP53 mutation rates and the incidence cancer exponent, regardless of the geographical area analyzed; albeit, the association is relatively weak in the United States. This supports a role for elevated levels of this inactivated gene in the rise of cancer incidence with aging, independent of sex, which is probably linked to a tumor’s requirement for genomic instability to escape replicative senescence and to utilize glycolytic pathways. The current relationship found between TP53 mutations and cancer incidence does not explain the declining cancer rates in the very elderly: one possibility is that the p53 gene exhibits antagonistic pleiotropy, suppressing cancer at the cost of accelerated aging.Citation57,Citation58 TP53 mutations probably account for about one-quarter of the aging-related increase in all cancers worldwide, and an even larger proportion of the aging-related increase in Japan, where obesity is uncommon. Therefore, productive avenues of future research include improving the consistency and decreasing the uncertainty of the TP53/cancer data analyzed, and determining by multivariant analysis whether a major part of the steep rise in cancer incidence with aging involves obesity or deficiencies in one of the many genes that are part of the TP53 signaling pathway.
Materials and Methods
TP53 mutations in cancer incidence and mortality data
Three large data sets were employed to provide epidemiologic data on cancer incidence or mortality, and TP53 mutations. The IARC GLOBOCAN Cancer DatabaseCitation59 provides ASR data (2008) in 5 y age groups for adults, covering 27 cancer sites in total for males, females, and both sexes. For United States incidence ASR data, I used SEER cancer statisticsCitation60 in addition to the GLOBOCAN database. The percentage of cancer samples with TP53 mutations for the cancers studied was obtained from the IARC TP53 Mutation Database, R15 version, November 2010.Citation3,Citation61
The prevalence of TP53 mutations worldwide for the incidence of all cancers (n = 25) and for solid tumors (n = 23) was calculated for both sexes as the sum of the product of different cancers’ TP53 mutation rates and ASR, divided by the total ASR.
Regional TP53 mutations and cancer incidence analyses
Analyses were performed for four geographical regions: worldwide, Japan, Western Europe, and the United States. The latter three regions were chosen because of the availability of TP53 mutation data (15 000–20 000 tumors sampled). Tumors were identified in the TP53 database () mainly by their anatomical site (topography), but also by their histology (morphology).
The cancer exponent for each of the cancers was calculated from the linear regression analysis of the log-transformed incidence or mortality ASR as the dependent variable and the log-transformed median age of 6 consecutive 5 y age groups as the independent variable. A small allowance of 5 y was made for lag between incidence and mortality ASRs. Accordingly, the cancer exponents were determined from 40–44 to 65–69 y of age for incidence ASR and from 45–49 to 70–74 y of age for mortality ASR.
Additional linear regression analyses were then conducted by region ri, i.e., r1–r4, for each of the cancer sites n with the percentage of samples with TP53 mutations as the independent variable and the cancer exponent as the dependent variable, i.e., TP53(n)ri vs. K(n)ri. Although there are large differences in age-dependent ASR () and in lifetime accumulated cancer rates for males and females, I did not account for sex differences in TP53 mutations. There is little evidence of sex differences in TP53 mutations human cancers, although there are exceptions, such as a higher rate of TP53 mutations in lung cancer among female smokers than among males.Citation21
The same analysis was conducted for all cancers and for solid tumors. Cancer sites for which there are less than 50 tumors were analyzed for TP53 mutations in the IARC TP53 Mutation Database were excluded, on the recommendation of the database manager. For each geographical region examined, the number of cancer sites with adequate samples varied, making the included sites for all cancers and for solid tumors variable by region. For all cancers, the highest number of sites was 25 (n). Analyses were first conducted for males and females for all cancers and followed by both sexes for all cancer and solid tumors, including OPC or SRT and excluding OPC or SRT. Prostate cancer, which had a regression standardized residual of ≥3.0, was identified as an outlier.
In addition, the slopes and means of the analysis of TP53 mutations vs. cancer exponents for each pair of regions (e.g., Japan:Western Europe) were compared by ANCOVA.
TP53 mutation rates and cancer incidence data by country
Finally, I examined for each country N whether the cancer exponent value K(N)ni for a specific cancer ni was influenced by the country TP53 mutation rate, i.e., TP53(N)ni. This relationship was assessed for 5 major cancer sites n1–n5: brain, breast—females only, colorectum, liver, and lung) for which the IARC TP53 Mutation Database has worldwide data from >5000 tumors for each cancer site. Prostate cancer was also assessed, although it is represented by only 1400 samples in the IARC TP53 Mutation Database accessing the R16 version. Countries with 50 or more tumor samples in the TP53 database for the cancer site were included.
As well, the relationship between TP53 mutations and cancer exponent, i.e., TP53(N)Ti vs. K(N)Ti was studied for the principal tumor Ti of the 5 sites (T1–T5: glioblastoma NOS, carcinoma NOS—females only, adenocarcinoma NOS, hepatocellular carcinoma NOS, and non-small cell carcinoma, respectively). IARC TP53 data for cancer sites was initially found by topography, whereas the TP53 data on principal tumors was found by their morphology.Citation3,Citation61
Body mass index and cancer incidence data by country
A non-TP53 influence, obesity, was briefly studied by use of age-standarized, mean body mass index (kg m−2) data by country provided by Imperial College London, School of Public HealthCitation20 for male and female adults in the year 2008. For prostate cancer, the number of countries was arbitrarily extended from 5 countries (with adequate TP53 data) to 24 countries by adding countries with adequate breast cancer TP53 data.
| Abbreviations: | ||
| ANCOVA | = | analysis of covariance |
| ASR | = | age-standardized rate |
| IARC | = | International Agency for Cancer Research |
| CI | = | confidence interval |
| OPC | = | outlier prostate cancer |
| SD | = | standard deviation |
| SEER | = | Surveillance, Epidemiology and End Results |
| SRT | = | sex-related tumors |
Acknowledgments
Wayne S Kendal of Ottawa University, Canada, is thanked his advice on statistical analysis.Citation62 Bruce McKay of Carleton University, Canada, and Nick Priest of AECL, Canada, provided helpful comments on the paper. I am grateful to Fred van Winckel, Statistics Canada, Ottawa, for providing data on cancer incidence and mortality in Canada. Magali Olivier of IARC, Lyon, France, is thanked for her aid in using the IARC TP53 database and comments on early drafts of the paper. David Malkin of The Hospital for Sick Children, Toronto is acknowledged for suggesting analysis of TP53 mutation rates from epithelial/non-epithelial cancers. Carolyn Brown of Ottawa is thanked for assistance in editing the paper. This study has benefitted from the library facilities that McGill University makes available to its adjunct professors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358:15 - 6; http://dx.doi.org/10.1038/358015a0; PMID: 1614522
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253:49 - 53; http://dx.doi.org/10.1126/science.1905840; PMID: 1905840
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat 2002; 19:607 - 14; http://dx.doi.org/10.1002/humu.10081; PMID: 12007217
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002; 415:45 - 53; http://dx.doi.org/10.1038/415045a; PMID: 11780111
- Rodier F, Campisi J, Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res 2007; 35:7475 - 84; http://dx.doi.org/10.1093/nar/gkm744; PMID: 17942417
- Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle 2007; 6:1006 - 10; http://dx.doi.org/10.4161/cc.6.9.4211; PMID: 17457049
- Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344 - 52; PMID: 20606252
- Boyle JO, Hakim J, Koch W, van der Riet P, Hruban RH, Roa RA, et al. The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res 1993; 53:4477 - 80; PMID: 8402617
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445:661 - 5; http://dx.doi.org/10.1038/nature05541; PMID: 17251932
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656 - 60; http://dx.doi.org/10.1038/nature05529; PMID: 17251933
- Chilosi M, Doglioni C, Menestrina F, Montagna L, Rigo A, Lestani M, et al. Abnormal expression of the p53-binding protein MDM2 in Hodgkin’s disease. Blood 1994; 84:4295 - 300; PMID: 7994045
- Goffin JR, Chappuis PO, Bégin LR, Wong N, Brunet JS, Hamel N, et al. Impact of germline BRCA1 mutations and overexpression of p53 on prognosis and response to treatment following breast carcinoma: 10-year follow up data. Cancer 2003; 97:527 - 36; http://dx.doi.org/10.1002/cncr.11080; PMID: 12548593
- Stubbert LJ, Smith JM, Hamill JD, Arcand TL, McKay BC. The anti-apoptotic role for p53 following exposure to ultraviolet light does not involve DDB2. Mutat Res 2009; 663:69 - 76; http://dx.doi.org/10.1016/j.mrfmmm.2009.01.010; PMID: 19428372
- Canadian Cancer Society’s Steering Committee on Cancer Statistics. (2011). Canadian Cancer Statistics 2011. (Toronto, ON: Canadian Cancer Society).
- Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer 1954; 8:1 - 12; http://dx.doi.org/10.1038/bjc.1954.1; PMID: 13172380
- Althuis MD, Dozier JM, Anderson WF, Devesa SS, Brinton LA. Global trends in breast cancer incidence and mortality 1973-1997. Int J Epidemiol 2005; 34:405 - 12; http://dx.doi.org/10.1093/ije/dyh414; PMID: 15737977
- Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 2009; 8:2031 - 40; http://dx.doi.org/10.4161/cc.8.13.8814; PMID: 19440038
- Hainaut P, Wiman KG. 30 years and a long way into p53 research. Lancet Oncol 2009; 10:913 - 9; http://dx.doi.org/10.1016/S1470-2045(09)70198-6; PMID: 19717093
- Ghartimagar D, Naik R, Gupta A, Ghosh A. A. Histopathology of prostatic lesions – an autopsy study of 100 cases. The Internet Journal of Forensic Science 2012; 5; http://dx.doi.org/10.1016/j.ccr.2010.09.005; PMID: 20951946
- Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, et al, Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (Body Mass Index). National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet 2011; 377:557 - 67; http://dx.doi.org/10.1016/S0140-6736(10)62037-5; PMID: 21295846
- Toyooka S, Tsuda T, Gazdar AF. The TP53 gene, tobacco exposure, and lung cancer. Hum Mutat 2003; 21:229 - 39; http://dx.doi.org/10.1002/humu.10177; PMID: 12619108
- Hill AB. The environment and disease: association or causation?. Proc R Soc Med 1965; 58:295 - 300; PMID: 14283879
- Höfler M. The Bradford Hill considerations on causality: a counterfactual perspective. Emerg Themes Epidemiol 2005; 2:11; http://dx.doi.org/10.1186/1742-7622-2-11; PMID: 16269083
- Malkin D. Li-Fraumeni syndrome. Genes Cancer 2011; 2:475 - 84; http://dx.doi.org/10.1177/1947601911413466; PMID: 21779515
- Jerry DJ, Ozbun MA, Kittrell FS, Lane DP, Medina D, Butel JS. Mutations in p53 are frequent in the preneoplastic stage of mouse mammary tumor development. Cancer Res 1993; 53:3374 - 81; PMID: 8324748
- Gao H, Wang LD, Zhou Q, Hong JY, Huang TY, Yang CS. p53 tumor suppressor gene mutation in early esophageal precancerous lesions and carcinoma among high-risk populations in Henan, China. Cancer Res 1994; 54:4342 - 6; PMID: 8044781
- Morgan C, Jenkins GJ, Ashton T, Griffiths AP, Baxter JN, Parry EM, et al. Detection of p53 mutations in precancerous gastric tissue. Br J Cancer 2003; 89:1314 - 9; http://dx.doi.org/10.1038/sj.bjc.6601302; PMID: 14520466
- Petitjean A, Achatz MIW, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene 2007; 26:2157 - 65; http://dx.doi.org/10.1038/sj.onc.1210302; PMID: 17401424
- Matoba S, Kang J-G, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science 2006; 312:1650 - 3; http://dx.doi.org/10.1126/science.1126863; PMID: 16728594
- Sood AK, Skilling JS, Buller RE. Ovarian cancer genomic instability correlates with p53 frameshift mutations. Cancer Res 1997; 57:1047 - 9; PMID: 9067268
- Shahedian B, Shi YF, Zou MJ, Farid NR. Thyroid carcinoma is characterized by genomic instability: evidence from p53 mutations. Mol Genet Metab 2001; 72:155 - 63; http://dx.doi.org/10.1006/mgme.2000.3114; PMID: 11161841
- Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002; 1:289 - 98; http://dx.doi.org/10.1016/S1535-6108(02)00047-8; PMID: 12086865
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:215 - 21; http://dx.doi.org/10.1038/356215a0; PMID: 1552940
- Mitchel RE, Jackson JS, Morrison DP, Carlisle SM. Low doses of radiation increase the latency of spontaneous lymphomas and spinal osteosarcomas in cancer-prone, radiation-sensitive Trp53 heterozygous mice. Radiat Res 2003; 159:320 - 7; http://dx.doi.org/10.1667/0033-7587(2003)159[0320:LDORIT]2.0.CO;2; PMID: 12600234
- Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ 2001; 322:1528 - 32; http://dx.doi.org/10.1136/bmj.322.7301.1528; PMID: 11420276
- Mai S, Hanley-Hyde J, Rainey GJ, Kuschak TI, Paul JT, Littlewood TD, et al. Chromosomal and extrachromosomal instability of the cyclin D2 gene is induced by Myc overexpression. Neoplasia 1999; 1:241 - 52; http://dx.doi.org/10.1038/sj.neo.7900030; PMID: 10935479
- Abulaiti A, Fikaris AJ, Tsygankova OM, Meinkoth JL. Ras induces chromosome instability and abrogation of the DNA damage response. Cancer Res 2006; 66:10505 - 12; http://dx.doi.org/10.1158/0008-5472.CAN-06-2351; PMID: 17079472
- Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 2008; 371:569 - 78; http://dx.doi.org/10.1016/S0140-6736(08)60269-X; PMID: 18280327
- Feng Z, Jin S, Zupnick A, Hoh J, de Stanchina E, Lowe S, et al. p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene 2006; 25:1 - 7; PMID: 16278683
- Chen Z, Trotman LC, Shaffer D, Lin H-K, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725 - 30; http://dx.doi.org/10.1038/nature03918; PMID: 16079851
- Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010; 18:367 - 81; http://dx.doi.org/10.1016/j.ccr.2010.09.005; PMID: 20951946
- Fulci G, Ishii N, Van Meir EG. p53 and brain tumors: from gene mutations to gene therapy. Brain Pathol 1998; 8:599 - 613; http://dx.doi.org/10.1111/j.1750-3639.1998.tb00187.x; PMID: 9804370
- Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med 2009; 361:2449 - 60; http://dx.doi.org/10.1056/NEJMra0804588; PMID: 20018966
- Zhu Y-M, Foroni L, McQuaker IG, Papaioannou M, Haynes A, Russell HH. Mechanisms of relapse in acute leukaemia: involvement of p53 mutated subclones in disease progression in acute lymphoblastic leukaemia. Br J Cancer 1999; 79:1151 - 7; http://dx.doi.org/10.1038/sj.bjc.6690183; PMID: 10098750
- Richardson RB. Ionizing radiation and aging: rejuvenating an old idea. Aging (Albany NY) 2009; 1:887 - 902; PMID: 20157573
- Richardson RB. Stem cell niches and other factors that influence the sensitivity of bone marrow to radiation-induced bone cancer and leukaemia in children and adults. Int J Radiat Biol 2011; 87:343 - 59; http://dx.doi.org/10.3109/09553002.2010.537430; PMID: 21204614
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396:643 - 9; http://dx.doi.org/10.1038/25292; PMID: 9872311
- Feng ZH, Hu WW, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci U S A 2007; 104:16633 - 8; http://dx.doi.org/10.1073/pnas.0708043104; PMID: 17921246
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153 - 8; http://dx.doi.org/10.1038/nature05610; PMID: 17344846
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science 2011; 331:1553 - 8; http://dx.doi.org/10.1126/science.1204040; PMID: 21436442
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108 - 13; http://dx.doi.org/10.1126/science.1145720; PMID: 17932254
- Shepard JL, Amatruda JF, Finkelstein D, Ziai J, Finley KR, Stern HM, et al. A mutation in separase causes genome instability and increased susceptibility to epithelial cancer. Genes Dev 2007; 21:55 - 9; http://dx.doi.org/10.1101/gad.1470407; PMID: 17210788
- Donehower LA. Genetic instability in animal tumorigenesis models. Cancer Surv 1997; 29:329 - 52; PMID: 9338107
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446:758 - 64; http://dx.doi.org/10.1038/nature05690; PMID: 17344859
- Richardson RB. Promotional etiology for common childhood acute lymphoblastic leukemia: the infective lymphoid recovery hypothesis. Leuk Res 2011; 35:1425 - 31; http://dx.doi.org/10.1016/j.leukres.2011.07.023; PMID: 21903265
- Gazdar AF, Butel JS, Carbone M. SV40 and human tumours: myth, association or causality?. Nat Rev Cancer 2002; 2:957 - 64; http://dx.doi.org/10.1038/nrc947; PMID: 12459734
- Harding C, Pompei F, Lee EE, Wilson R. Cancer suppression at old age. Cancer Res 2008; 68:4465 - 78; http://dx.doi.org/10.1158/0008-5472.CAN-07-1670; PMID: 18519710
- Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle 2010; 9:3151 - 6; http://dx.doi.org/10.4161/cc.9.16.13120; PMID: 20724817
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893 - 917; http://dx.doi.org/10.1002/ijc.25516; PMID: 21351269
- Fast Stats: An interactive tool for access to SEER cancer statistics Bethesda MSRP, National Cancer Institute.
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007; 28:622 - 9; http://dx.doi.org/10.1002/humu.20495; PMID: 17311302
- Porter AMW. Misuse of correlation and regression in three medical journals. J R Soc Med 1999; 92:123 - 8; PMID: 10396255