Abstract
The inflammation regulating transcription factor NFκB and the tumor-suppressing transcription factor p53 can act as functional antagonists. Chronic inflammation (NFκB activity) may contribute to the development of cancer through the inhibition of p53 function, while, conversely, p53 activity may dampen inflammation. Here we report that the E3 ubiquitin ligase MDM2, whose gene is transcriptionally activated by both NFκB and p53, can bind and inhibit the p65RelA subunit of NFκB. The interaction is mediated through the N-terminal and the acidic/zinc finger domains of MDM2 on the one hand and through the N-terminal Rel homology domain of p65RelA on the other hand. Co-expression of MDM2 and p65RelA caused ubiquitination of the latter in the nucleus, and this modification was dependent of a functional MDM2 RING domain. Conversely, inhibition of endogenous MDM2 by small-molecule inhibitors or siRNA significantly reduced the ubiquitination of ectopic and endogenous p65RelA. MDM2 was able to equip p65RelA with mutated ubiquitin moieties capable of multiple monoubiquitination but incapable of polyubiquitination; moreover, MDM2 failed to destabilize p65RelA detectably, suggesting that the ubiquitin modification of p65RelA by MDM2 was mostly regulatory rather than stability-determining. MDM2 inhibited the NFκB-mediated transactivation of a reporter gene and the binding of NFκB to its DNA binding motif in vitro. Finally, knockdown of endogenous MDM2 increased the activity of endogenous NFκB as a transactivator. Thus, MDM2 can act as a direct negative regulator of NFκB by binding and inhibiting p65RelA.
Introduction
The dimeric transcription factor nuclear factor kappa B (NFκB) is activated by various danger signals to orchestrate the immune response and to control the survival, proliferation, and differentiation of cells.Citation1 In the canonical NFκB pathway in resting cells, stress receptors stimulate NFκB kinase (IKK) to phosphorylate the inhibitors of NFκB (IkBs), typically IkBα, which is then ubiquitinated and degraded through the proteasome to liberate NFκB from cytoplasmic sequestration.Citation1,Citation2 Nuclear NFκB regulates a plethora of genes until newly translated IkBα shuttles NFκB back to the cytoplasm or until NFκB is ubiquitinated and degraded.Citation3,Citation4 The canonical transactivating NFκB is a heterodimer consisting of p65RelA and p50. p65RelA is regulated by posttranslational modifications, including ubiquitination which may reduce its DNA binding capacity and stability.Citation5,Citation6 The E3 ubiquitin ligases PDLIM2 and SOCS1/COMMD1 are known as p65RelA-modifying enzymes.Citation7-Citation9
In contrast to NFκB, the homotetrameric transcription factor and tumor suppressor p53 is activated mostly by endogenous stresses such as DNA damage and the inappropriate activity of oncogenes, usually resulting in cell senescence or apoptosis through the stimulation of genes, including the p21Waf/Cip1 cell cycle arrest gene and the pro-apoptotic PUMA gene.Citation10 p53’s activity in cells is limited by the multifunctional, mono- or oligomeric, nuclear and partly cytoplasmic E3 ubiquitin ligase MDM2.Citation11-Citation13 MDM2, whose gene is transactivated by p53, acts as a central negative regulator of p53 at basically three levels: the ubiquitin-marking for degradation of p53, the export of p53 from the nucleus, and the direct transcriptional repression of promoters recognized by p53.Citation14-Citation19
Recent intriguing discoveries document functional antagonism of NFκB and p53 in at least some settings.Citation20,Citation21 For example, while NFκB typically transactivates pro-proliferative and anti-apoptotic genes, p53 often transactivates anti-proliferative and pro-apoptotic genes.Citation22 Moreover, NFκB and p53 engage in reciprocal negative regulation, i.e., NFκB activity can suppress p53 response and vice versa.Citation23-Citation26 The underlying mechanisms include competition for limiting cofactors, such as p300,Citation27-Citation29 IKK-mediated degradation of p53,Citation30 and the functional antagonism of products of NFκB- and p53-responsive genes.Citation31 First hints that MDM2 may also have a role in this reciprocal interaction included the observation that both NFκB and p53 transactivate the MDM2 gene,Citation32-Citation34 and that MDM2, in turn, stimulates the p65RelA promoter and increases p53 expression by interacting with p53’s mRNA.Citation35,Citation36 Altogether, in contrast to the inhibition of p53 by NFκB, the mechanisms underlying the inhibition of NFκB by p53 are less well defined. Here we show that MDM2 can bind to p65RelA and inhibit its function.
Results
MDM2 binds NFκB subunit p65RelA
A previous search for proteins that associate with the RING-type E3 ubiquitin ligase MDM2 had indicated subunit p65RelA of the heterodimeric transcription factor NFκB as a potential binding partner. To confirm the interaction, initially GST pulldown assays were performed with full-length MDM2 fused N-terminally to GST as the bait, and with in vitro-translated 35S-labeled full-length p65RelA as the prey. While GST alone failed to retain p65RelA, GST-MDM2 bound and coprecipitated it (). Next we studied the interaction of the two proteins in vivo. For this purpose, human H1299 lung adenocarcinoma cells deficient for p53 were transfected to express combinations of plasmids producing p65RelA and MDM2. p53-deficient cells were chosen because p53 can transactivate the endogenous MDM2 gene and can bind to both MDM2 and p65RelA.Citation11-Citation13,Citation28,Citation37 At 24 h after transfection, cell lysates were incubated with monoclonal anti-p65RelA, anti-MDM2, or irrelevant antibody and standard immunoprecipitates were analyzed by western blotting. As summarized in (left panel), anti-p65RelA antibody precipitated p65RelA and coprecipitated ectopic MDM2, whereas irrelevant antibody did not. Conversely, precipitation of ectopic MDM2 coprecipitated p65RelA (, right panel). Moreover, a fraction of the endogenous MDM2 present in human p53-deficient HCT116 colon adenocarcinoma cells coprecipitated with endogenous p65RelA (). Finally, to obtain information on the interaction domains, full-length p65RelA or N-terminal and C-terminal fragments of p65RelA, each with a Flag-tag, were cotransfected with full-length MDM2, and coprecipitation of MDM2 with Flag-p65RelA was studied. shows that the N-terminal 310 aa residues of Flag-p65RelA containing the Rel homology domain (RHD) bound strongly to MDM2, whereas the C-terminal half of the protein (aa 311–550) containing the two transactivation domains failed to bind. In accord with previous observations,Citation38 the C-terminus of p65RelA was less well expressed than the N-terminus in vivo. To identify the MDM2 domain that contacts p65RelA, full-length MDM2 or fragments of MDM2 were coexpressed with p65RelA, and coprecipitations were again analyzed by western blotting. p65RelA efficiently coprecipitated full-length MDM2 protein as well as MDM2 fragment 6–339 containing the N-terminal p53-binding domain and the central acidic and zinc finger (A/Z) domains. In contrast, p65RelA coprecipitated MDM2 delta222–325 lacking the A/Z domains much weaker than full-length MDM2 or MDM2 6–339 (). The C-terminus, including the RING domain of MDM2, was dispensable for the binding of p65RelA. Thus, p65RelA associates with the A/Z domains and the N-terminus of MDM2. MDM2 mutant D68A that is defective for the efficient binding of p53 readily bound p65RelA (not shown). Combined, these data indicate that the N-terminal Rel homology domain of NFκB subunit p65RelA binds to the central acidic and zinc finger domains, and to the N-terminus, of MDM2.
Figure 1. MDM2 binds p65RelA in vitro and in vivo. (A) GST pulldown assay. In vitro -translated, 35S-labeled p65RelA is retained by bacterially expressed GST-MDM2 but not GST alone. The lower panel shows the expression of the GST proteins in bacteria. One-tenth of the labeled p65RelA was used as input control. (B) Coimmunoprecipitation of transfected proteins. Human H1299 cell cultures were transfected with expression plasmids producing p65RelA (p65; 4 μg) and/or MDM2 (4 μg). At 24 h after transfection, cells were permeabilized and the extracts were incubated with the indicated polyclonal (p65) or monoclonal (MDM2) antibodies to immunoprecipitate p65 (left panel) or MDM2 (right panel). Immunoprecipitation (IP) samples, co-immunoprecipitation (Co-IP) samples, and total cell lysates (TCL) were probed by western immunoblotting with antibodies directed against p65, MDM2, and β-actin. IgG was an irrelevant control antibody. (C) Co-immunoprecipitation of endogenous proteins. Human HCT116 p53−/− cell cultures were treated with TNFα (20 ng/ml) for 2 h prior to permeabilization. Endogenous p65RelA (p65) was precipitated with anti-p65 polyclonal antibody. Immunoprecipitation (IP) samples, co-immunoprecipitation (Co-IP) samples, and total cell lysates (TCL) were probed by western immunoblotting with the indicated antibodies. IgG was an irrelevant control antibody.
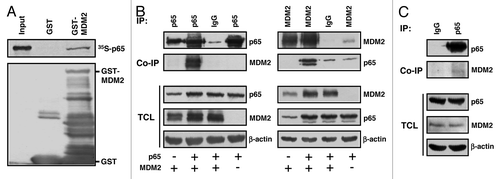
Figure 2. Analysis of the protein interaction domains. H1299 cells were transfected for 24 h with plasmids producing the indicated Flag-tagged fragments of p65RelA plus MDM2 (A) or with plasmids expressing the depicted MDM2 fragments plus full-length p65 (B). Cell extracts were incubated with anti-Flag or anti-p65 antibodies to immuno/co-immunoprecipitate the transfected proteins, and the precipitates were analyzed by western blotting with the indicated antibodies. Note that Flag-p65-Cterm was only weakly expressed (A, lane 4). RHD, Rel homology domain; TAD, transactivation domain; p53, p53-binding domain; A/Z, acidic and zinc finger domains; R, RING domain.
MDM2 ubiquitinates p65RelA
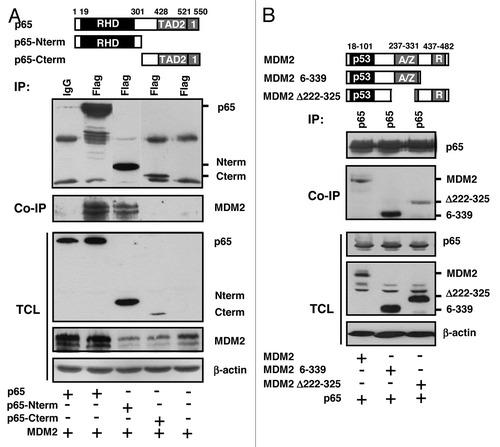
MDM2 functions as a RING-type ubiquitin ligase to a large number of cellular proteins, among them p53. Immunofluorescence microscopy of cell cultures transfected with Flag-p65RelA and HA-MDM2 showed that ectopic Flag-p65RelA is localized in the cytoplasm as well as in the cell nucleus, whereas ectopic HA-MDM2 is predominantly nuclear (). To begin to investigate if MDM2 can ubiquitinate p65RelA, plasmids expressing Flag-p65RelA, MDM2, and HA-tagged ubiquitin were cotransfected. Flag-p65RelA was immunoprecipitated from cell extracts prepared under denaturing conditions, as reported previously,Citation39 in order to avoid detecting ubiquitinated coprecipitating proteins instead of ubiquitinated Flag-p65RelA. HA-ubiquitinated Flag-p65RelA was detected by western immunoblotting. The results document that a fraction of Flag-p65RelA was ubiquitinated in the presence of ectopic MDM2, and that this ubiquitination was dependent of the level of transfected MDM2 (, lanes 2–5). RING domain mutant G448S/C449A of MDM2 known to be defective for ubiquitination bound to Flag-p65RelA as efficiently as wild-type MDM2 () but failed to ubiquitinate Flag-p65RelA above background (, compare lanes 2, 7 and 8), suggesting that ubiquitination was direct and not secondary, i.e., through another MDM2-interacting ubiquitin ligase. Preparation of cytoplasmic and nuclear fractions from cell cultures transfected with different combinations of Flag-p65RelA- and MDM2-expression plasmids indicated that the ubiquitination of Flag-p65RelA by MDM2 was taking place in the nucleus (), the compartment where both ectopic proteins primarily localized. Note that MDM2 was expressed in , lane 4 because Flag-p65RelA transactivated the endogenous MDM2 gene.
Figure 3. Ectopic MDM2 ubiquitinates p65RelA. (A) Colocalization of Flag-p65 and HA-MDM2. H1299 cells were transfected with 0.2 μg of Flag-tagged p65RelA and 0.2 μg of HA-tagged MDM2. Immunofluorescence signals were produced 24 h after transfection by incubation of the fixated and permeabilized cells with FITC-conjugated anti-Flag and TRITC-conjugated anti-HA antibodies. The merged images show that both proteins can colocalize in the nucleoplasm in a fraction of the transfected cells. (B) Wild-type MDM2 but not MDM2 mutant G448S/C449A HA-ubiquitinates Flag-p65. H1299 cells were transfected with the indicated combinations of plasmids expressing Flag-p65 (1 μg), MDM2 (0.1, 0.2, 0.5 μg) or MDM2-G448S/C449A (0.5 μg), and HA-ubiquitin (3 μg), for 24 h. All cultures were then incubated in the presence of the proteasome inhibitor MG132 (10 μM) for another 4 h, after which the cells were lysed under denaturing conditions, and Flag-p65 was immunoprecipitated with anti-Flag antibody. Immunoprecipitates (IP) and total cell lysates (TCL) were analyzed by western immunoblotting with antibodies directed against Flag-p65, MDM2 and β-actin. (C) Wild-type MDM2 and MDM2 mutant G448S/C449A bind to Flag-p65 with equal efficiency. H1299 cells were transfected with expression plasmids for Flag-p65 (4 μg), MDM2 (4 μg) or MDM2-G448S/C449A (4 μg; MDM2*) as indicated. At 24 h after transfection, Flag-p65 was immunoprecipitated from cell extracts and co-precipitating MDM2 was analyzed by western blotting. Cell lysates were prepared under denaturing conditions. HA-ubiquitinated protein species were identified by western blotting with anti-HA antibody. IP, immunoprecipitation; TCL, total cell lysate.
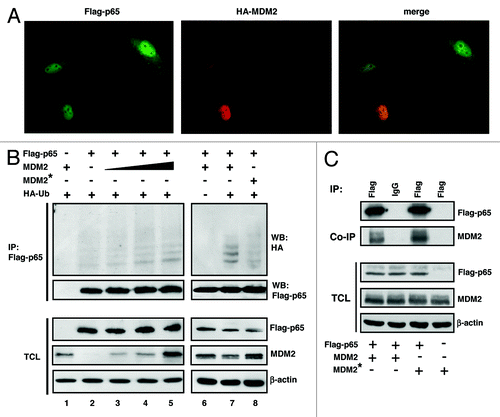
Figure 4. Ubiquitination of p65RelA by MDM2. (A) MDM2 is predominantly nuclear in H1299 cells, and ubiquitination of Flag-p65 is taking place in the cell nucleus. Cultures were transfected with the indicated expression plasmids (Flag-p65, 1 μg; MDM2, 1 μg), and cells were fractionated at 24 h after transfection into a cytoplasmic and nuclear fraction (CF and NF), respectively. The quality of the fractionation was monitored by the immunodetection of a cytoplasmic protein (α-tubulin; 56 kDa) and a nuclear protein (pRB; 110 kDa). Ubiquitinated Flag-p65 protein species appeared as a characteristic high-molecular weight smear (80 to > 250 kDa) in western immunoblots. (B) p65RelA is ubiquitinated by wild-type HA-tagged ubiquitin and equally efficiently by HA-tagged ubiquitin mutants K48R (left panel) and K3xR (= K29,48,63R, right panel) that are impaired (K48R) and incapacitated (K3xR) for polyubiquitination. H1299 cells were transfected for 24 h with plasmids producing the indicated proteins (HA-ubiquitin wild-type, HA-ubiquitin K48R, and HA-ubiquitin K3xR, 3 μg each; p65, 3 μg; MDM2, 1 μg), and all cultures were incubated with the proteasome inhibitor MG132 (10 μM) for another 4 h. Cell lysates were prepared under denaturing conditions. p65RelA was immunoprecipitated, and the HA-ubiquitinated protein species were identified by western blotting with anti-HA antibody. IP, immunoprecipitation; TCL, total cell lysate.
Ubiquitination of transfected p65RelA by transfected MDM2 seemed to preferentially cause an increase in the signal intensity of individual slow migrating bands indicative of multiple lysine monoubiquitination (see, for example, ). Multiple monoubiquitination of proteins is well known to regulate protein function rather than degradation through the 26S proteasome, whereas the formation of polyubiquitin chains with more than four ubiquitin molecules covalently linked to lysine 48 of ubiquitin seems to constitute the major signal for proteasomal protein degradation.Citation40 To test whether MDM2 can monoubiquitinate p65RelA, cells were transfected with combinations of plasmids producing p65RelA, MDM2, and either a wild-type HA-ubiquitin or a HA-ubiquitin incapacitated for polyubiquitination due to a K48R mutation. MDM2 was able to link HA-ubiquitin-K48R to p65RelA as efficiently as HA-ubiquitin wild-type (). Since it was known that ubiquitin residues K29 and K63, in addition to K48, may be employed for polyubiquitination, we next made use, in a similar experiment, of an ubiquitin that had all three lysines mutated (K3xR). Again, MDM2 caused ubiquitination of p65RelA with this ubiquitin mutant as efficiently as with wild-type ubiquitin (). These findings thus suggest that p65RelA can be multiply monoubiquitinated by MDM2. Consistent with these observations and with previous reports that p65RelA is highly stable in a large number of different cell lines,Citation41-Citation43 we have not been able to detect an effect of ectopic MDM2 on the half-life of p65RelA (not shown).
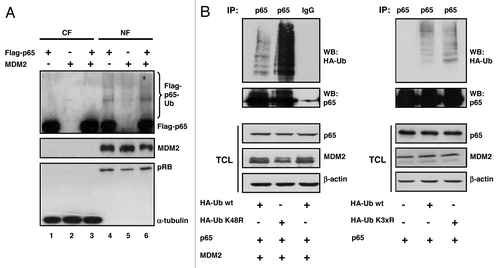
Consistent through all ubiquitination experiments, some Flag-p65RelA was ubiquitinated even in the absence of ectopic MDM2 (see, for example, , lane 2 and , lane 2). We hypothesized that this was due at least in part to the activity of endogenous MDM2. In another experimental approach we therefore initially asked if the p65RelA binding-competent but ubiquitination-defective mutant MDM2-G448S/C449A would function in a dominant-negative manner if overproduced. Toward this end, cotransfections of plasmids expressing Flag-p65RelA, HA-ubiquitin, and either wild-type MDM2 or MDM2-G448S/C449A were performed, and the HA-ubiquitination of Flag-p65RelA immunoprecipitated from denatured cell extracts was examined by western blotting. As visible in , overproduced MDM2-G448S/C449A not only was inactive for catalyzing the ubiquitination observed in the presence of ectopic MDM2 but inhibited background ubiquitination (compare lanes 2 and 6), suggesting that this p65RelA-binding mutant (see ) was able to interfere with p65RelA modification by endogenous ubiquitin ligase(s) in a dominant-negative manner. To gain information on the effect of endogenous MDM2 on transfected p65RelA, we next expressed p65RelA and HA-ubiquitin in the presence and absence of two chemical inhibitors of MDM2. Inhibitor I (N-[{3,3,3-Trifluoro-2-trifluoromethyl}propionyl]sulphanilamide) is a cell-permeable, simple reversible inhibitor that does not prevent substrate binding and is highly selective for the E3 ligase activity of MDM2.Citation44 Inhibitor II (HLI373) (5-[3-Dimethylaminopropylamino)-3,10-dimethyl-10H-pyrimido[4,5-b]quinoline-2,4-dione,2HCl) is a cell-permeable selectively MDM2-inhibiting pyrimidoquinoline-dione.Citation45 The experiment showed that HA-ubiquitination was completely abrogated in the presence of a cocktail of inhibitor I and II compared with mock treatment (). Interestingly, incubation of cells with the inhibitor cocktail for 24 h resulted in a strong reduction specifically of the MDM2 protein levels as detected by antibodies 3G9 and 4B11, whose epitope recognition capabilities are known to be insensitive to posttranslational modification.Citation46 To corroborate the suggestion that the observed HA-ubiquitination of p65RelA primarily occurred through endogenous MDM2, we studied, in a further experiment, the ubiquitination of endogenous p65RelA upon siRNA-mediated depletion of MDM2. As with the inhibitors of MDM2, siRNA-mediated knockdown of MDM2 transcript resulted in the disappearing of the short-lived MDM2 protein and in the cessation of p65RelA background HA-ubiquitination (). Of note, treatment of cells with the MDM2 inhibitors I and II also resulted in a dose-dependent reduction of p65RelA ubiquitination with the polyubiquitination-defective ubiquitin mutant K3xR (), suggesting that endogenous MDM2 can monoubiquitinate p65RelA at multiple lysine residues. Combined, these results thus indicate that the NFκB subunit p65RelA is substrate for ubiquitination by the E3 ubiquitin ligase MDM2.
Figure 5. Ubiquitination of p65RelA by endogenous MDM2. (A) Ubiquitination-defective MDM2 mutant G448S/C449A inhibits ubiquitination of p65RelA by endogenous ubiquitin ligases. H1299 cells were transfected with combinations of plasmids producing the indicated proteins (Flag-p65, 1 μg; MDM2, 0.1, 0.2 or 0.5 μg; MDM2* [= MDM2 G448S/C449A], 0.5 μg; HA-Ubiquitin, 3 μg). Twenty-four h after transfection, cells were treated as described in the legend to , and Flag-p65 was immunoprecipitated with anti-Flag monoclonal antibody from denatured cell extracts. The levels of HA-ubiquitination and of protein expression were determined by standard western blotting using the antibodies directed against the indicated proteins. (B) Correlation of the level of expression of endogenous MDM2 with the HA-ubiquitination of p65RelA upon treatment of cultures with small-molecule MDM2 inhibitors. Cultures of H1299 cells were transfected for 24 h with plasmids expressing p65RelA (3 μg) and HA-ubiquitin (3 μg). Cells were then either mock treated or treated with a cocktail of inhibitor I (200 μM) and inhibitor II (5 μM) for 24 h, and were finally exposed to the proteasome inhibitor MG132 (10 μM) for 4 h. Note that the low levels of endogenous MDM2 (H1299 cells are p53-deficient), as detected by the MDM2 antibodies 3G9 and 4B11, decreased further after drug exposure. Immunoprecipitation of p65RelA and the detection of HA-ubiquitination and protein expression were again done as outlined in the legend to . (C) siRNA recognizing MDM2 transcript but not scrambled control siRNA caused a knockdown of MDM2 expression and concomitantly, cessation of HA-ubiquitination of endogenous p65RelA. H1299 cells were transfected with HA-ubiquitin producing plasmid for 24 h, and were then RNAi-fected with the indicated siRNAs (c = control; mdm2) for further 24 h. Finally, cells were treated with MG132 (10 μM) for 4 h and were subjected to p65RelA immunoprecipitation as before. Proteins were detected with the indicated antibodies in standard Western immunoblottings. (D) Linking of monoubiquitination-proficient ubiquitin moieties to p65RelA by endogenous ubiquitin ligase is reduced in the presence of MDM2 inhibitors. H1299 cells were transfected with plasmids producing p65RelA and ubiquitin mutant K3xR (K29,48,63R) impaired for polyubiquitination for 24 h, and were then treated with MG132 (10 μM) as well as, where indicated, with increasing quantities of MDM2 inhibitors I (50, 125, 250 μM) and II (1, 2.5, 5 μM) for another 4 h. Analysis of ubiquitination was as in B. IP, immunoprecipitation; WB, western blot.
![Figure 5. Ubiquitination of p65RelA by endogenous MDM2. (A) Ubiquitination-defective MDM2 mutant G448S/C449A inhibits ubiquitination of p65RelA by endogenous ubiquitin ligases. H1299 cells were transfected with combinations of plasmids producing the indicated proteins (Flag-p65, 1 μg; MDM2, 0.1, 0.2 or 0.5 μg; MDM2* [= MDM2 G448S/C449A], 0.5 μg; HA-Ubiquitin, 3 μg). Twenty-four h after transfection, cells were treated as described in the legend to Figure 1B, and Flag-p65 was immunoprecipitated with anti-Flag monoclonal antibody from denatured cell extracts. The levels of HA-ubiquitination and of protein expression were determined by standard western blotting using the antibodies directed against the indicated proteins. (B) Correlation of the level of expression of endogenous MDM2 with the HA-ubiquitination of p65RelA upon treatment of cultures with small-molecule MDM2 inhibitors. Cultures of H1299 cells were transfected for 24 h with plasmids expressing p65RelA (3 μg) and HA-ubiquitin (3 μg). Cells were then either mock treated or treated with a cocktail of inhibitor I (200 μM) and inhibitor II (5 μM) for 24 h, and were finally exposed to the proteasome inhibitor MG132 (10 μM) for 4 h. Note that the low levels of endogenous MDM2 (H1299 cells are p53-deficient), as detected by the MDM2 antibodies 3G9 and 4B11, decreased further after drug exposure. Immunoprecipitation of p65RelA and the detection of HA-ubiquitination and protein expression were again done as outlined in the legend to Figure 1B. (C) siRNA recognizing MDM2 transcript but not scrambled control siRNA caused a knockdown of MDM2 expression and concomitantly, cessation of HA-ubiquitination of endogenous p65RelA. H1299 cells were transfected with HA-ubiquitin producing plasmid for 24 h, and were then RNAi-fected with the indicated siRNAs (c = control; mdm2) for further 24 h. Finally, cells were treated with MG132 (10 μM) for 4 h and were subjected to p65RelA immunoprecipitation as before. Proteins were detected with the indicated antibodies in standard Western immunoblottings. (D) Linking of monoubiquitination-proficient ubiquitin moieties to p65RelA by endogenous ubiquitin ligase is reduced in the presence of MDM2 inhibitors. H1299 cells were transfected with plasmids producing p65RelA and ubiquitin mutant K3xR (K29,48,63R) impaired for polyubiquitination for 24 h, and were then treated with MG132 (10 μM) as well as, where indicated, with increasing quantities of MDM2 inhibitors I (50, 125, 250 μM) and II (1, 2.5, 5 μM) for another 4 h. Analysis of ubiquitination was as in B. IP, immunoprecipitation; WB, western blot.](/cms/asset/481c3de3-2aa4-492e-a0ac-28029f78a24a/kccy_a_10925495_f0005.gif)
MDM2 inhibits NFκB-mediated gene transactivation
NFκB with its subunit p65RelA is an important inducible transcription factor that becomes activated in response to various extracellular stresses. Next we therefore explored the involvement of MDM2 in the regulation of NFκB-stimulated transcription. In a first set of experiments, human Jurkat T cells that are widely used for transient NFκB-reporter studies, as they express only minute levels of active p65RelA, were transiently cotransfected with combinations of plasmids expressing Flag-p65RelA and MDM2 plus a reporter plasmid containing a luciferase gene driven by five copies of a κB-recognition sequence. As expected, Flag-p65RelA strongly transactivated the luciferase reporter gene compared with a plasmid that lacked the κB-motifs (). Notably, increasing levels of MDM2 expression plasmid significantly reduced this transactivation, consistent with MDM2 acting as an inhibitor of NFκB-mediated gene activation. However, it was known that MDM2 can act as a general, unspecific inhibitor of transactivation by directly interacting with elements of the basal transcription machinery.Citation18 To exclude the possibility that in these experiments MDM2 had reduced the expression of luciferase simply by inhibiting the Flag-p65RelA plasmid, we monitored the transcript levels by RT-PCR. The Flag-p65RelA transcript level was not affected by the presence of MDM2 (). Thus, MDM2 inhibited Flag-p65RelA-induced transactivation of an NFκB-responsive luciferase gene in this reporter gene assay.
Figure 6. MDM2 inhibits NFκB-mediated gene transactivation. (A) Ectopic MDM2 represses reporter gene activation by ectopic p65RelA. Human Jurkat T cells were transiently transfected with reporter plasmid NFκB-luc harbouring a NFκB-responsive luciferase gene (0.2 μg) plus the indicated combinations of plasmids producing either Flag-p65RelA (0.1 μg) or MDM2 (+, 0.05 μg; ++, 0.5 μg). At 24 h after transfection, cell extracts were prepared for luciferase assay. The bar diagram shows the luciferase activity over background (reporter plasmid only; arbitrarily chosen as 1). T-bars denote the standard deviations derived from three experiments. The P value was calculated by Student t test (two-tailed). Below the diagram are the results of standard RT-PCRs for the expression of the Flag-p65RelA, MDM2 and GAPDH transcripts in the transfected cultures. Note that increasing MDM2 expression does not inhibit the expression of Flag-p65RelA. (B) Endogenous p65RelA activated by TNFα is inhibited by ectopic MDM2. Human 293T cells that are routinely used for the study of TNFmediated NFκB activation were transfected with the NFκB-sensitive reporter plasmid plus empty vector as control or plasmid expressing MDM2 or MDM2-G448S/C449A (MDM2*; +, 0.05 μg; ++, 0.2 μg; +++, 0.5 μg). In addition, cultures were either mock treated or exposed to TNFα (20 ng/ml) for 6 h to induce nuclear translocation of the cytoplasmic NFκB. Cell extracts were prepared for luciferase assay 30 h after transfection. T bars indicated standard deviations derived from six experiments. The P value was calculated by Student t test (two-tailed). (C) Pharmacological inhibition of MDM2 by small-molecule inhibitors increases NFκB activity. 293T cells were transfected with the NFκB reporter plasmid as before and then mock treated or treated with a cocktail of MDM2 inhibitors I (200 μM) and II (5 μM) for another 24 h. All cultures either received TNFα (20 ng/ml) or not, for another 6 h. Luciferase activity is expressed as fold activation over background (reporter plasmid with mock treatment). Standard deviations were derived from six experiments; P values (*) were determined by Student t test (two-tailed). (D) Knockdown of MDM2 by siRNA increases NFκB activity. 293T cells were transfected with the reporter and then RNAi-fected with either control siRNA (c) or MDM2 siRNA for another 24 h. Cultures were treated with TNFα as before. MDM2 knockdown was monitored by western immunoblotting. The standard deviations were calculated from six experiments, and the P value (*) was determined by Student t test (two-tailed). (E) Knockdown of MDM2 by siRNA increases expression of endogenous NFκB-responsive genes. 293T cultures were RNAi-fected for 24 h and exposed to TNFα as in (C and D). Total RNA was prepared, reversely transcribed and analyzed by RT-qPCR for the transcript levels of the indicated genes relative to gapdh level. P values were determined as before.
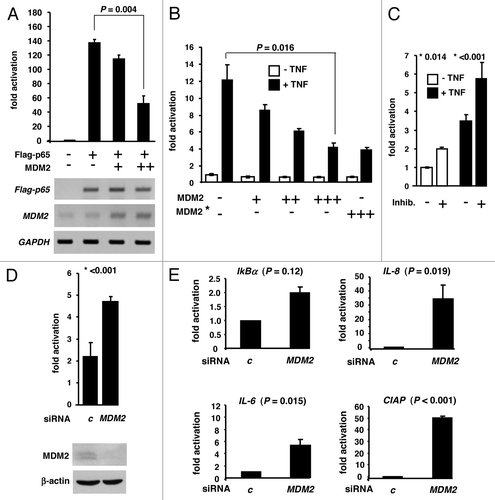
Human 293T cells contain cytoplasmic NFκB that is translocated to the nucleus upon treatment with TNFα. Accordingly, TNFα exposure of cultures transfected with the NFκB-responsive reporter plasmid resulted in robust luciferase activity. Again, this activity was inhibited by cotransfected MDM2 plasmid in a dose-dependent manner (). Consistent with previous findings on p53,Citation18 the inhibition of NFκB by MDM2 was not dependent of the ubiquitin ligase activity. Ligase-defective yet binding-competent MDM2 mutant G448S/C449A proved to be suppressive in this assay (), suggesting that binding of MDM2 was sufficient for NFκB inhibition. Conversely, when the endogenous MDM2 was inhibited by the small-molecule MDM2 inhibitors I and II () or was knocked down by MDM2 siRNA (), the NFκB-mediated transactivation of the reporter increased significantly. To validate enhanced NFκB activity in 293T cultures depleted for MDM2, we next studied, by RT-qPCR, the effect of MDM2 knockdown on the expression of endogenous NFκB-responsive genes. As documented in , MDM2 knockdown resulted in the increased transcription of the NFκB-stimulated IkBα gene relative to gapdh, although this increase did not reach statistical significance. However, MDM2 depletion caused robust and significant upregulation of the NFκB target genes IL-8, IL-6, and CIAP. A similar but less pronounced effect was observed in the absence of TNFα stimulation, suggesting that even in the absence of stimulation, some NFκB was active as a transcription factor and was controlled by MDM2 (not shown). In sum, these results thus indicate that ectopic as well as endogenous MDM2 can inhibit NFκB-mediated gene activation.
MDM2 inhibits the DNA binding of NFκB in vitro
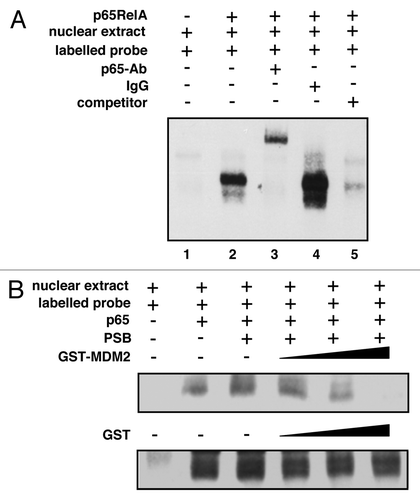
Since MDM2 associated with the DNA-binding Rel homology domain (RHD) of p65RelA (see ), it was conceivable that MDM2 can interfere with the DNA binding of NFκB. To examine the effect of MDM2 on NFκB DNA binding in vitro, nuclear extracts from HeLa cells transfected to produce p65RelA were employed in standard electrophoretic mobility shift assays (EMSA) with a labeled NFκB binding motif as probe. p65RelA expression induced the formation of a slowly migrating complex in non-denaturing PAGE (). Consistent with this complex containing p65RelA, the band could be specifically supershifted by a monoclonal antibody directed against p65RelA but not by an irrelevant IgG. Moreover, complex formation could be inhibited by the incubation with excess non-labeled probe. To test the effect of MDM2 on the formation of the complex, increasing quantities of purified GST-MDM2 or GST alone was included in the EMSA. While GST failed to affect the DNA binding activity of NFκB, GST-MDM2 reduced it in a dose-dependent manner (). Similar results were obtained with extracts from H1299 cells (data not shown). These findings thus suggest that MDM2 can block the DNA binding activity of NFκB by interacting with p65RelA.
Figure 7. MDM2 can inhibit the DNA binding activity of NFκB. (A) NFκB with ectopic p65RelA binds and retards a biotin-labeled oligonucleotide bearing an NFκB DNA binding motif. EMSA was performed with nuclear extracts prepared from HeLa cells that had been transfected either with control plasmid or with a plasmid expressing p65RelA. The extracts were mock incubated or incubated with monoclonal anti-p65RelA antibody C-20 (p65-Ab) or an irrelevant IgG to assay for the presence of p65RelA in the complex. In addition, the sample in lane 5 was incubated with a 200-fold excess of unlabelled NFκB DNA binding motif (competitor) to rule out band shifting due to interaction with biotin. (B) Increasing amounts of GST-MDM2 but not GST reduce the DNA binding activity of NFκB. EMSA was performed as in (A), except that the extracts were mixed with increasing amounts of purified GST-MDM2 or GST (0.1, 0.2 or 0.4 μg), where indicated. Protein storage buffer (PSB), either with or without GST or GST-MDM2 protein, was added to equal concentrations.
p53 can induce p65RelA ubiquitination with dependence on MDM2
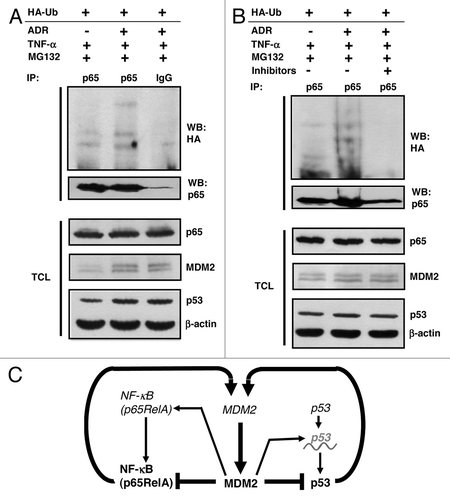
DNA damage typically causes a delayed increase of the endogenous MDM2 level in cells with an intact wild-type p53 pathway, since active p53 transactivates the MDM2 gene. Next, we therefore asked whether p53 activation by the radiomimetic DNA-damaging drug doxorubicin can affect the ubiquitination of p65RelA in a MDM2-dependent manner. To this end, human p53-proficient HCT116 colon adenocarcinoma cells were transfected to produce HA-ubiquitin and were either mock treated or treated with doxorubicin (0.34 μM) for 10 h. The cultures were then exposed to TNFα (to translocate NFκB to the nucleus) and MG132 for another 2 h. As expected, p53 was activated by doxorubicin and the level of MDM2 was elevated (, lower panels). Incubation of the denatured cell extracts with polyclonal anti-p65 antibody or irrelevant IgG immunoprecipitated p65RelA only in the presence of specific antibody. The subsequent analysis for HA-ubiquitination revealed a correlation of p65RelA ubiquitination and MDM2 level, consistent with p65RelA being ubiquitinated by MDM2 under these conditions (). To corroborate this suggestion, MDM2 was inhibited by the small-molecule inhibitors I and II in a similar experiment. MDM2 inhibition completely abrogated this effect (). It should be noted that p65RelA ubiquitination in response to doxorubicin treatment was not detectable when the cells were exposed to TNFα and MG132 for 6 h instead of only 2 h. The reason for this discrepancy has not been elucidated. However, it is conceivable that p65RelA and MDM2 after 6 h no longer colocalize and/or that they receive modifications that disrupt their interaction. In conclusion, MDM2 can act as a novel direct negative regulator of NFκB by binding and inhibiting p65RelA. summarizes some of the p53:MDM2:p65RelA feedback interactions.
Figure 8. Regulation of p65RelA by p53 via MDM2. (A) Increase of p65RelA ubiquitination in cells overproducing endogenous MDM2 upon activation of endogenous p53. HCT116 cells harboring wild-type p53 were transfected with 4 μg of plasmid producing HA-ubiquitin. After 24 h, cultures were treated with doxorubicin (0.34 μM) for 10 h to activate p53. Finally, cells were exposed to TNFα (20 ng/ml) and MG132 (10 μM) for another 2 h. HA-ubiquitinated p65RelA was immunoprecipitated from denatured cell extracts with polyclonal anti-p65RelA antibody or irrelevant antibody (IgG). Proteins were analyzed by standard western blotting. TCL, total cell lysate. (B) Same experiment as in (A), except that one of the cultures that were activated with doxorubicin received, in addition to TNFα and MG132, a cocktail of the MDM2 inhibitors I (200 μM) and II (5 μM) for 2 h. (C) Schematic presentation of some of the MDM2:p65RelA and MDM2:p53 feedback interactions. Genes are shown in italic, proteins in bold print. p53 (italic, with wave line) indicates p53 mRNA. Arrows denote activation, T bars inhibition.
Discussion
Danger-sensing cell surface receptors are crucial elements of the innate and adaptive immune system. They frequently activate the cytoplasmic canonical NFκB transcription factor heterodimer consisting of the transactivation-domain containing p65RelA and transcriptionally inactive p50 to translocate to the nucleus and stimulate numerous genes whose products mediate inflammation.Citation1,Citation2 Obviously, this inflammatory response has to be halted after the danger vanished, and it is thus no surprise that a number of mechanisms has evolved to terminate NFκB activity, among them protein/protein interactions, phosphorylation, and ubiquitination.Citation4 Polyubiquitination of p65RelA seems to be primarily catalyzed by the ubiquitin ligases SOCS1/Cullin-2/COMMD1 and PDLIM2 and can occur through the growth of polyubiquitin chains at the lysine residues 29, 33, 48, and 63 of ubiquitin. Polyubiquitination via K48 typically constitutes the signal for the degradation of p65RelA by the 26S proteasome.Citation5,Citation7,Citation8
Recent work has documented that in addition to degradation-inducing polyubiquitination, p65RelA is subject to monoubiquitination at multiple lysine residues, including K62, K123, and K315.Citation47 This monoubiquitination is taking place in the nucleus and, in contrast to polyubiquitination, does not affect protein stability. Instead it results in the nuclear sequestration of transcriptionally inactive p65RelA, perhaps through the inhibition of its interaction with IκBα that would cause shuttling to the cytoplasm. The responsible nuclear monoubiquitin ligase(s) is unknown. We have shown here that the N-terminal and the acidic/zinc finger domains of the E3 ubiquitin ligase MDM2 can bind to the N-terminal Rel homology domain (RHD) of p65RelA. Binding of the ubiquitin ligase SOCS1/Cullin-2/COMMD1 to p65RelA also involves the RHDCitation5 and has been suggested to be inhibited by phosphorylation of the RHD at serine residues 205, 276, and 281.Citation47,Citation48 Whether the interaction of MDM2 with p65RelA is regulated by phosphorylation of the RHD as well is unknown. However, such regulation would be consistent with our data showing that only a fraction of p65RelA was ubiquitinated by MDM2, and that this fraction could be increased only slightly by the expression of further ectopic MDM2. It is thus conceivable that modification of the RHD prevents the interaction of MDM2 and p65RelA and/or redirects p65RelA to a different cellular sub-compartment to limit the amount of it that is accessible to MDM2.
MDM2 may poly- and/or monoubiquitinate p65RelA. Our data obtained with the polyubiquitination-impaired, monoubiquitination-proficient ubiquitin mutants K48 and K3xR (K29,48,63R) suggest that MDM2 can efficiently link monoubiquitin to multiple lysine residues of p65RelA. However, we also observed that the level of ubiquitinated p65RelA in the presence of MDM2 could be enhanced by the proteasome inhibitor MG132, which would be consistent with MDM2 polyubiquitin-marking p65RelA for proteasome-mediated degradation. On the other hand, and consistent with previous reports,Citation41-Citation43 p65RelA was stable in all cell lines tested, compared with other targets of MDM2-marked degradation such as p53 and MDM2 itself, and MG132 treatment could not detectably stabilize it further. As a possible solution for this seeming contradiction, we would like to suggest that the effect of MG132 does not primarily involve p65RelA stabilization. We observed that MDM2 itself was stabilized in response to MG132 treatment, leading to greatly increased MDM2 levels, and we therefore suggest that this, rather than the inhibited breakdown of p65RelA, caused the increased levels of ubiquitinated p65RelA that was observed in the presence of the drug. This of course provokes the question why we failed to observe that overproduced ectopic MDM2 mimics MG132 treatment. One reason could be that proteasome inhibition by MG132 constitutes a danger signal that leads to abrogation of inhibitory posttranslational modifications of the p65RelA RHD to facilitate MDM2/p65RelA interaction. Future work will address this possibility. We believe that the ubiquitination of p65RelA by MDM2 regulates NFκB’s function rather than its stability, a suggestion that is in accord with previous findings. Inhibition of p65RelA phosphorylation at S205, S276, and S281 within the RHD supported monoubiquitination in the cell nucleus and transcriptional inhibition of NFκB, independent of protein degradation.Citation47 Nuclear MDM2-mediated p65RelA inhibition may thus be part of a fail-safe mechanism that could become activated in the case that the major inhibitor of NFκB, IκBα, is non-functional and proteasome-mediated protein degradation is compromised.
NFκB binds to specific DNA sequences through the RHDs of its subunits. Several proteins have been identified that can inhibit this DNA binding by associating with the RHD of p65RelA. For example, the transcriptional regulator and E3 SUMO ligase PIAS1 (protein inhibitor of activated STAT1) can associate with the RHD of p65RelA and inhibit its DNA binding.Citation38 Here we report that the E3 ubiquitin ligase MDM2 can also contact the RHD of p65RelA and inhibit the transcriptional activator function of NFκB. Like PIAS1, MDM2 impeded the binding of NFκB to the κB-DNA motif in vitro. It is unclear at present whether this effect is caused by DNA contact inhibition and/or the inhibition of p65RelA:p50 dimer formation. Other NFκB-regulating factors such as the E3 ubiquitin ligase SOCS1 (suppressor of cytokine signaling 1) can interact with DNA-bound NFκB and terminate its activity by ubiquitin-marking it for proteasomal degradation.Citation6,Citation9 MDM2 was able to ubiquitinate p65RelA, and previous work has shown that the addition of a single ubiquitin moiety to the N-terminus of p65RelA was sufficient to impair transcriptional activity of NFκB.Citation47 However, and consistent with MDM2:RHD contact impeding DNA binding, ubiquitination was not a prerequisite for NFκB inhibition, as the ubiquitination-incompetent yet p65RelA binding-competent MDM2 mutant G448S/C449A nonetheless was a formidable transcriptional inhibitor. Other mechanisms through which MDM2 may retard transcription include the blocking of coactivator recruitment and the association of MDM2 with the 34 kDa subunit of TFIIE of the basal transcription machinery.Citation14-Citation18 It may thus be that, depending on context, MDM2 can inhibit NFκB through at least three mechanisms: (1) ubiquitination; causing degradation, cellular translocation, or impairment of DNA binding, (2) direct impairment of DNA binding, perhaps involving the regulation of NFκB subunit dimer formation and affecting only a subset of the NFκB responsive genes, and (3) the recruitment of MDM2 to NFκB-bound promoters.
NFκB and the tumor suppressor p53 are functional antagonists in at least some contexts, and MDM2 may be a major player in this relation. Since both NFκB and p53 can transactivate the MDM2 gene,Citation32-Citation34 the inhibition of either by MDM2 must be regulated in a manner that permits reciprocal control. In factual terms, if the impairment of p53 transcriptional activity by NFκB stimulating cytokinesCitation23-Citation25 and conversely, the suppression of NFκB by p53Citation49,Citation50 are at least in part MDM2-dependent, the p53:MDM2 and NFκB:MDM2 interactions should be under the reign of specific posttranslational modifications that regulate function or cause spaciotemporal separation of the protagonists. We showed here that MDM2 accumulation in response to p53 activation can lead to enhanced p65RelA ubiquitination, but only under the specific condition of short-term proteasome inhibition. Unraveling the precise role of MDM2 in the entangled p53-NFκB pathway will be a challenge. For example, MDM2 depletion by RNAi technology will not only affect NFκB in this pathway, but will cause accumulation of active p53 which itself can bind and affect NFκB, independent of MDM2.Citation37 Perhaps the development of small-molecule inhibitors that are highly specific for the disruption of the MDM2:p65RelA interaction can help solve this dilemma.
To characterize such inhibitors, combined systems biological and network modeling approaches complemented by biochemical analyses may be helpful, as has been shown recently, for example, for the MDM2 inhibitor of the spiro-oxindole family, MI-219.Citation51 MI-219 disrupts the MDM2-p53 interaction, thereby increasing cellular p53 levels; however, MDM2 inhibition by MI-219 also increased the cellular levels of NFκB subunit p65RelA. Moreover, microarray and Ingenuity pathway analyses revealed activation of the NFκB central hub as the primary effect of MI-219 treatment after 16 h. It is thus conceivable that small-molecule inhibitors of MDM2 can stimulate NFκB through affecting the MDM2:p65RelA interaction identified here. Of note, at later times of MI-219 treatment (32 h) the NFκB hub, although retained as a central pathway responding to MI-219, was now suppressed by MDM2 inhibitionCitation51 suggesting that, in turn, cellular contexts exist in which MDM2 activates rather than suppresses NFκB as would follow from the results presented here. Such a cellular response, leading to the activation of NFκB by MDM2 independent of p53, has indeed recently been reported in the context of acute kidney injury.Citation52 Clearly, these issues await further analysis.
Crosstalk between NFκB and p53 is implemented on multiple levels, not only in the cell nucleus. For example, the cytoplasmic NFκB-activating enzymes IKKα/β induce the MDM2-mediated degradation of p53, while cIAP2, a cellular inhibitor of apoptosis, can block this mechanism by supporting the SUMOylation of MDM2.Citation53 In turn, pharmacological inhibition of p53 causes IKK activation and, consequently, the activation of NFκB. Interestingly, this activation seems to require the function of two genes involved in (macro)autophagy, the lysosomal digestion of macromolecular cellular structures in response to starvation.Citation54 It is unclear at present whether (cytoplasmic) MDM2 has a mediating role in this mechanism as well. Such a role could involve, besides the function as ubiquitin ligase, the activity of MDM2 as a cytoplasmic stabilizer and inducer of translation of mRNAs. For instance, MDM2 binds and regulates the translation not only of p53’s mRNACitation36 but also of the mRNA of the MYCN transcription factor, which, like NFκB, seems to be linked to p53 by substantial crosstalk.Citation55 So does cytoplasmic MDM2 also regulate NFκB pathway-relevant mRNAs? Future work will tell.
Materials and Methods
Plasmids, chemicals, and antibodies
T7-RelA, pCMV4-p65, pcDNA3-HA-Ubiquitin, pRK5-HA-Ubiquitin-K48R were purchased from Addgene. GST-MDM2 full-length was generated by PCR and cloned into pGEX-4T1 (Amersham). Flag-p65 full-length and Flag-p65 deletion mutants were also produced by PCR and were subcloned into pcDNA3.1(+)-Neo (Invitrogen). Cloning details are available upon request. MDM2 mutants G448S/C449A, 6–339 and ∆222–325 were kindly provided by Matthias Dobbelstein (Department of Molecular Oncology, Georg-August–Universität Göttingen) and pcDNA3.1-Ha-Ubiquitin (K29, 48, 63R) by Ivan Dikic (Institute of Biochemistry II, Goethe University). The reporter gene vectors pCIS-CK and pNFκB-Luc were purchased from Agilent Technologies. Aprotinin (# A1153), MG132 (# M7449), doxorubicin (# D1515, adriamycin), PMSF (# P7626), protease inhibitor cocktail (# P8345), and 4’,6-Diamidin-2’-phenylindoldihydrochlorid (DAPI, # D9542) were from Sigma, as were the β-actin monoclonal antibody (clone AC-15, # A5441), the monoclonal anti-FLAG M2 antibody (# F3165), the monoclonal anti-glutathione-s-transferase antibody (clone GST-2, # G1160), the monoclonal anti-FLAG M2-FITC (clone M2, # F4049), the monoclonal anti-HA-TRITC (clone HA-7, # H9037), and the peroxidase-conjugated secondary anti-mouse (# A9044) and anti-rabbit antibodies (# A6154) as well as all oligonucleotides. The polyclonal anti-NFκB p65-antibody (# ab7970) and the monoclonal anti-α-tubulin-antibody (clone DM1A, # ab7291) were purchased from Abcam; anti-Mdm2-antibody 3G9 (# 04–1530) was from Millipore. The irrelevant monoclonal anti-HRS3-antibody as well as the polyclonal anti-TGF-β receptor-antibody were kindly provided by Michael Pfreundschuh (Internal Medicine I, University of Saarland Medical Center). Transfection reagent Nanofectin I (# Q051–005) was from PAA (Pasching, Austria), RNAifect (# 301605) from Qiagen. MDM2 E3 ligase inhibitor I (N-[{3,3,3-Trifluoro-2-trifluromethyl}propionyl] sulphanilamide; # 373225) and II (5-[3-Dimethylaminopropylamino-3,10-dimethyl-10H-pyrimido{4,5-b}quinoline-2,4-dione; # 373226]) were purchased from Calbiochem as were the anti-MDM2-antibody 4B11 (# OP143) and the anti-p53-antibody DO-1 (# OP43L). Monoclonal anti-HA-antibody (HA.11 clone 16B12, # MMS-101P) was from Covance. TNFα (# 210-TA/CF) was purchased from R&D Systems. The monoclonal anti-Rb-antibody (clone G3–245, #554136) was from BD Biosciences. BSA (#1.12018.0100) was purchased from Merck.
Cell culture and transfection
All cells were maintained at 37 °C in a 7% CO2 atmosphere. H1299 cells and HEK293T were maintained in DMEM (#E15–843, PAA) with 10% fetal calf serum (#A15–101, PAA). HCT116 cells were maintained in McCoy 5A Medium (#M9309, Sigma) with 10% fetal calf serum. For transient transfection, cells were seeded to reach 60–70% confluency at transfection and were transfected with Nanofectin I (PAA) following to the manufacturer’s recommendations.
GST pulldown assay
For GST pulldown analyses, equal amounts of GST and GST-MDM2 were immobilized on Gluthation-Sepharose beads (# 17-0756-01, GE-Healthcare), washed 5 times with GST low salt buffer (50 mM Tris/HCl, 200 mM NaCl, 0.8 mM EDTA, 0.1% NP40, 1 mM PMSF, 10 µg/ml aprotinin) and incubated with equal amounts of p65RelA protein (in vitro-translated from T7-RelA). The in vitro translation was performed with the TNT-T7 Coupled Reticulocyte Lysate System (# L4610), according to the manufacturer’s protocol (Promega), with 1 µg plasmid and 35S radiolabeled cysteine and methionine (Tran35S-Label™, # 51006, MP Biomedicals). After overnight incubation at 4 °C, all probes were washed 5 times with GST high-salt buffer (50 mM Tris/HCl, 500 mM NaCl, 0.8 mM EDTA, 0.1% NP40). GST protein complexes were eluted from the sepharose by adding SDS-sample buffer (100 mM TRIS-HCl [pH 6.8], 100 mM DTT, 4% SDS, and 20% glycerol) and by boiling samples for 10 min. The proteins were separated by SDS-PAGE, immobilized on PVDF membrane (Immobilon P, # IPVH00010, Millipore) and visualized by autoradiography. GST-proteins were detected with the monoclonal anti-glutathione-s-transferase antibody (clone GST-2).
Co-immunoprecipitation
H1299 cells were seeded to ~60% confluency and were transfected with the indicated plasmids with Nanofectin I (PAA) following to the manufacturer’s recommendations. After 24 h, the cells were treated with 10 µM MG132 (Sigma) for 2 to 6 h. Four µg of the indicated antibodies were added to a 1:1:1 mixture of protein A sepharose 4 Fast Flow (#17-5280-01), protein G sepharose 4 Fast Flow (#17-0618-01) and γ-Bind-sepharose (#17-0885-01, all GE-Healthcare) in co-IP lysis buffer (0,5% NP-40, 150 mM NaCl, 20 mM Na2HPO4 × 12 H2O, 5 mM EDTA, 10% glycerol supplemented with freshly added 1 mM PMSF and protease inhibitors) and incubated for at least 4 h on a rotating wheel at 4 °C. Unbound antibody was removed by 2–3× washing with co-IP lysis buffer. Cells were harvested and washed in sterile phosphate buffer saline. 1/10 of the cells was lysed in SDS-sample buffer (100 mM TRIS-HCl [pH 6.8], 100 mM DTT, 4% SDS, and 20% glycerol) and served as an input control. The remaining cells were permeabilized for 30 min on ice in co-IP lysis buffer. The extracts were then centrifuged at 16 000 g for 30 min at 4 °C. The supernatant was incubated with the sepharose-antibody-mix for at least 4 h at 4 °C on a rotating wheel. Precipitates were washed three times with co-IP lysis buffer before SDS-sample buffer was added to the sepharose beads. All probes were boiled for 10 min and subjected to SDS-PAGE for western blot analysis. For the co-IP of endogenous proteins, HCT116 p53−/− were seeded to 90% confluency and treated with TNFα (20 ng/ml) for 2 h prior harvesting.
Protein extraction and western blot analysis
Cells were lysed in SDS-lysis buffer heated to 100 °C, containing 100 mM TRIS-HCl (pH 6.8), 100 mM DTT, 4% SDS, and 20% glycerol. Fifteen µg to 30 µg protein were subjected to 8–13% SDS-PAGE and transferred to a PVDF membrane (Immobilon-P; #IPVH00010, Millipore). Signals were detected upon overnight incubation of the membranes with one of the indicated antibodies (α-GST 1:1.000; α-NFκB p65 1:1.000, α-β-actin 1:10.000, α-Mdm2 clone 3G9 1:2.000, α-Flag 1:10.000, α-HA 1:1.000, α-Mdm2 clone 4B11 1:1.000; α-p53 DO-1 1:2.000; α-tubulin 1:10.000, α-Rb 1:500), followed by a further incubation with a peroxidase-conjugated secondary anti-mouse (1:2.000) or anti-rabbit (1:2.000) antibody, and detected by the Thermo Scientific ECL Western Blotting substrate (#32106) as specified by the supplier.
MDM2 knockdown
Short interfering RNA (Hs_MDM2_5- sense: UCAUCGGACUCAGGUACAUTT; antisense: AUGUACCUGAGUCCGAUGATT) was used to silence gene expression. As a control, an irrelevant siRNA (control- sense: r(UUCUCCGAACGUGUCACGU)dTdT; antisense r(ACGUGACACGUUCGGAGAA)dTdT) was used. All siRNAs were purchased from QIAGEN. Exponentially growing H1299 or HEK293T cells were transfected with siRNA (40 nM) by RNAifect (Qiagen).
Immunofluorescence analyses
0.3 × 105 H1299 cells were seeded in 4-chamber polystyrene vessel tissue culture treated glass slides (#354114, BD Falcon), and 24 h after seeding they were transfected with the indicated plasmids. Cells were fixed in paraformaldehyde (4% in PBS), permeabilized on ice with 0.2% Triton-X/PBS for 2 min, blocked for 30 min with 1 µg/ml BSA (Merck) at 37 °C and were then incubated with anti-Flag-FITC (1:200) or anti-HA-TRITC (1:200) or both, for 1 h. After several washing steps with PBS and 0.03% Triton-X/PBS, the nuclei (DNA) were stained with 0.2 µg/ml DAPI (4’,6-diamidin-2’-phenylindoldihydrochlorid, Sigma) in methanol. The slides were mounted with PermaFluorTM Aqueous mounting medium (#TA-006-FM, Thermo Scientific). A Leica DM IRB/E fluorescence microscope equipped with an Axio Cam color camera (Zeiss) was used for microscopy; data were analyzed with the Axio Vision 3.0 software.
In vivo ubiquitination of p65/RelA
H1299 cells were seeded in 10 cm dishes the day before transfection to reach a confluency of 70% to 80%. Cells were transfected with the indicated plasmids. After further 24 h, cells were treated with 10 µM MG132 for 4–6 h. Cells were then washed in cold PBS; one-tenth of the cells were saved as input control. The rest was lysed in 400 µl TBS-lysis buffer (1% SDS in TBS) per 10 cm dish at 95 °C for 5 min. Lysates were squeezed repeatedly through a 23-gauge needle and vortexed vigorously for 10 sec. Eight hundred µl TBS-Triton buffer (1.5% Triton X-100 in TBS) per 400 µl lysate was added and mixed prior to incubation with 100 µl of a 1:1 mix of protein G and protein A sepharose 4 Fast Flow (GE Healthcare) for 1 h on a rotating wheel at 4°C (preclearing). Samples were centrifuged for 5 min at max speed and supernatant was incubated with 100 µl of a 1:1 mix of protein G and protein A sepharose 4 Fast Flow preconjugated with 4 µg of the indicated antibody for at least 4 h at 4 °C on a rotating wheel. Samples were washed three times in 1 ml cold TBS mix (1 part TBS-lysis buffer plus 2 parts TBS-Triton buffer); beads were resuspended in 30 µl of 95 °C SDS-sample buffer (100 mM TRIS-HCl (pH 6.8), 100 mM DTT, 4% SDS, and 20% glycerol) and were boiled for 10 min. The proteins were separated by SDS-PAGE, immobilized on PVDF membrane (Immobilon P, Millipore) and detected by the indicated antibodies.
RT-PCR and RT-qPCR
Cells were seeded in 10 cm, 6 cm, or 35 mm dishes and after 24 h were transfected and/or treated with the indicated reagents. At various times cells were lysed in solution D (236.4 g guanidium thiocyanate in 293 ml water, 17.6 ml 0.75 M sodium citrate pH 7.0, and 26.4 ml 10% sarcosyl, 0.72% 2-mercaptoethanol). Lysate was harvested and 0.1 ml of 2 M sodium acetate pH 4.0, 1 ml of water-saturated phenol (Roth), and 0.2 ml of chloroform-isoamylalcohol (49:1) were added, mixed, and cooled on ice for 15 min. After centrifugation (10 000 g, 20 min, 4 °C), the aqueous phase was collected and precipitated with isopropanol at −20 °C overnight. After a further centrifugation (10 000 g, 20 min, 4 °C), RNA was dissolved in solution D and precipitated with isopropanol at −20 °C for 1 h. The pellet was washed in 70% ethanol and dissolved in DEPC-water. The RNA was digested with RNase-free DNase I (#10776785001, Roche) for 60 min at 37 °C, and 1.5–5 µg was used for the first-strand cDNA synthesis with SuperScript™III (#18080-093, Invitrogen) as specified by the manufacturer. Semiquantitative RT-PCR analysis was performed with AmpliTaqR Gold DNA polymerase (#N808-0242, Applied Biosystems), using the following primers: Flag-p65 (for: gattataaagatgatgatgataaa; rev: ctacaagctcgtgggggtgaggcc; TA: 64 °C), MDM2 (for: atcgaatccggatcttgatg; rev: tcttgtccttcttcactaaggc; TA: 64 °C), gapdh (for: tggtatcgtggaaggactcatgac; rev: agtccagtgagcttcccgttcagc; TA: 64 °C). Quantitative RT-PCR analysis for IκBα, IL-8, IL-6, CIAP, and gapdh was performed with the LightCycler® FastStart DNA Master SYBR Green I from Roche (#12239264001) using the following primers: IκBα (for: gatccgccaggtgaaggg; rev: gcaatttctggctggttgg); IL-8 (for: atgacttccaagctggccgt; rev: ttacataatttctgtgttggc), IL-6 (for: aggcactggcagaaaacaac; rev: gaggtgcccatgctacattt); CIAP (for: ctgggaaccaaaggatgatg; rev: attggtgggtcagcattttc), gapdh (for: tggtatcgtggaaggactcatgac; rev: agtccagtgagcttcccgttcagc; TA: 64 °C); final concentration of primers: 0.5 µM; final MgCl2 concentration: 2 mM).
Reporter gene analysis
One × 105 Jurkat cells or 0.3 × 105 HEK293T cells were seeded in 24-well-dishes. After 24 h, cells were transfected with Nanofectin I with the plasmid combination described in the figure legends. After further 24 h, cells were either left untreated or were treated with a cocktail of MDM2 E3 ligase inhibitor I (200 µM) and II (5 µM), or were RNAi-fected with either control or MDM2 siRNA for further 24 h. Prior to harvesting, cells were treated with TNFα (20 ng/ml) or left untreated as specified in the figure legends. Luciferase assays were performed with the Luciferase Assay System (#1500, Promega) as specified by the manufacturer.
Fractionation of cells
H1299 cells were seeded to reach 80% confluency at time of transfection. After 24 h, cells were transfected with Nanofectin I with the plasmid combinations described in the figure legends. After another 24 h, cells were washed with ice-cold PBS, harvested and washed again with ice-cold PBS. One-fifth of the cells were saved as total cell extract and lysed in 1× SDS-lysis buffer (100 mM TRIS-HCl (pH 6.8), 100 mM DTT, 4% SDS, and 20% glycerol). The rest of the cells was centrifuged, resuspended in 300 µl ice-cold hypotonic buffer A (10 mM HEPES pH 7.9, 10 mM KCl, supplemented with freshly added 1 mM PMSF and protease inhibitors) and incubated on ice for 15 min. Fifteen µl NP-40 solution (10% NP-40 in H2O) was then added to reach a final concentration of 0.5%, and the samples were vortexed immediately. Samples were centrifuged for 5 min at 800 g; supernatant was the cytoplasmic fraction which was diluted 1:1 with 2× SDS-lysis buffer (200 mM TRIS-HCl [pH 6.8], 100 mM DTT, 8% SDS, and 40% glycerol) and boiled for 10 min at 95 °C. The pellet was the nuclear fraction; it was washed once with 1 ml of ice-cold buffer A plus NP-40 (final concentration 0.5%). The pellet was lysed in 1x SDS-lysis buffer and boiled for 10 min at 95 °C. The protein concentrations were determined and 15 or 30 µg of protein was analyzed by standard western immunoblotting. Anti-tubulin antibody was used as marker for the cytoplasmic fraction, anti-Rb antibody as a marker for the nuclear fraction.
Electrophoretic mobility shift assay
EMSA analyses were performed using the LightShift® Chemiluminescent EMSA Kit (#20148, Thermo Scientific). One µg of nuclear extracts were examined for their binding capacity to 20 fmol of the biotin-labeled standard NFκB binding motif (5′-AGCTTCAGAGGGGACTTTCCGAGAGG-3′).Citation56 Binding reaction and electrophoresis on a 4% native polyacrylamide gel were performed as recommended by the manufacturer. Complexes were then transferred onto the Hybond™-N+ nylon membrane (#RPN303B, GE Healthcare). For supershift analysis, we used 2 µg anti-NFκB p65RelA rabbit polyclonal antibody (C-20, #sc-372 X, Santa Cruz Biotechnology) or irrelevant polyclonal anti-TGF-β receptor-antibody (kindly provided by Michael Pfreundschuh, Internal Medicine I, University of Saarland Med Center) as control. For oligonucleotide competitor experiments, 4 pmol of unlabeled NFκB site were added to the binding reaction prior to incubation with the biotin-labeled probe. The biotin-labeled DNA was detected by chemiluminescence as described by the manufacturer. To examine the effect of MDM2 on NFκB DNA binding activity, the nuclear extracts were incubated for 30 min at room temperature with increasing amounts of purified GST-Mdm2 (#H00004193-P01, abnova) or alternatively, with GST alone (#P0001, abnova) as control.
| Abbreviations: | ||
| MDM2 | = | murine double minute 2 |
| NFκB | = | nuclear factor-kappa B |
| IκBα | = | inhibitor of kappa B alpha |
Acknowledgments
We thank Drs Ivan Dikic, Matthias Dobbelstein and Michael Pfreundschuh for materials. CW, FG, and CS are Master-of-Science students supported by the University of Saarland. This work was financed by a HOMFOR grant from the University of Saarland to KR.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest are disclosed.
Related Research Data
References
- Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012; 26:203 - 34; http://dx.doi.org/10.1101/gad.183434.111; PMID: 22302935
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol 2011; 12:695 - 708; http://dx.doi.org/10.1038/ni.2065; PMID: 21772278
- Smale ST. Hierarchies of NF-κB target-gene regulation. Nat Immunol 2011; 12:689 - 94; http://dx.doi.org/10.1038/ni.2070; PMID: 21772277
- Ruland J. Return to homeostasis: downregulation of NF-κB responses. Nat Immunol 2011; 12:709 - 14; http://dx.doi.org/10.1038/ni.2055; PMID: 21772279
- Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell 2003; 12:1413 - 26; http://dx.doi.org/10.1016/S1097-2765(03)00490-8; PMID: 14690596
- Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor kappaB response. J Exp Med 2004; 200:107 - 13; http://dx.doi.org/10.1084/jem.20040196; PMID: 15226358
- Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol 2007; 8:584 - 91; http://dx.doi.org/10.1038/ni1464; PMID: 17468759
- Maine GN, Mao X, Komarck CM, Burstein E. COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J 2007; 26:436 - 47; http://dx.doi.org/10.1038/sj.emboj.7601489; PMID: 17183367
- Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J 2011; 25:863 - 74; http://dx.doi.org/10.1096/fj.10-170597; PMID: 21084693
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8:275 - 83; http://dx.doi.org/10.1038/nrm2147; PMID: 17380161
- Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res 2003; 1:993 - 1000; PMID: 14707282
- Meek DW, Knippschild U. Posttranslational modification of MDM2. Mol Cancer Res 2003; 1:1017 - 26; PMID: 14707285
- Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 2010; 20:299 - 309; http://dx.doi.org/10.1016/j.tcb.2010.01.009; PMID: 20172729
- Barak Y, Gottlieb E, Juven-Gershon T, Oren M. Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev 1994; 8:1739 - 49; http://dx.doi.org/10.1101/gad.8.15.1739; PMID: 7958853
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387:296 - 9; http://dx.doi.org/10.1038/387296a0; PMID: 9153395
- Ohtsubo C, Shiokawa D, Kodama M, Gaiddon C, Nakagama H, Jochemsen AG, et al. Cytoplasmic tethering is involved in synergistic inhibition of p53 by Mdmx and Mdm2. Cancer Sci 2009; 100:1291 - 9; http://dx.doi.org/10.1111/j.1349-7006.2009.01180.x; PMID: 19432880
- Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev 1994; 8:1235 - 46; http://dx.doi.org/10.1101/gad.8.10.1235; PMID: 7926727
- Thut CJ, Goodrich JA, Tjian R. Repression of p53-mediated transcription by MDM2: a dual mechanism. Genes Dev 1997; 11:1974 - 86; http://dx.doi.org/10.1101/gad.11.15.1974; PMID: 9271120
- Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell 2004; 16:631 - 9; http://dx.doi.org/10.1016/j.molcel.2004.10.016; PMID: 15546622
- Gudkov AV, Gurova KV, Komarova EA. Inflammation and p53: A Tale of Two Stresses. Genes Cancer 2011; 2:503 - 16; http://dx.doi.org/10.1177/1947601911409747; PMID: 21779518
- Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol 2011; 12:715 - 23; http://dx.doi.org/10.1038/ni.2060; PMID: 21772280
- Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, et al. p53 is a suppressor of inflammatory response in mice. FASEB J 2005; 19:1030 - 2; PMID: 15811878
- Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991; 352:345 - 7; http://dx.doi.org/10.1038/352345a0; PMID: 1852210
- O’Prey J, Crighton D, Martin AG, Vousden KH, Fearnhead HO, Ryan KM. p53-mediated induction of Noxa and p53AIP1 requires NFkappaB. Cell Cycle 2010; 9:947 - 52; http://dx.doi.org/10.4161/cc.9.5.10872; PMID: 20160496
- Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med 1999; 190:1375 - 82; http://dx.doi.org/10.1084/jem.190.10.1375; PMID: 10562313
- Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci U S A 2005; 102:17448 - 53; http://dx.doi.org/10.1073/pnas.0508888102; PMID: 16287968
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A 1997; 94:2927 - 32; http://dx.doi.org/10.1073/pnas.94.7.2927; PMID: 9096323
- Ikeda A, Sun X, Li Y, Zhang Y, Eckner R, Doi TS, et al. p300/CBP-dependent and -independent transcriptional interference between NF-kappaB RelA and p53. Biochem Biophys Res Commun 2000; 272:375 - 9; http://dx.doi.org/10.1006/bbrc.2000.2786; PMID: 10833421
- Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell 2007; 26:75 - 87; http://dx.doi.org/10.1016/j.molcel.2007.02.019; PMID: 17434128
- Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci U S A 2009; 106:2629 - 34; http://dx.doi.org/10.1073/pnas.0812256106; PMID: 19196987
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 2007; 8:49 - 62; http://dx.doi.org/10.1038/nrm2083; PMID: 17183360
- Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell 2002; 1:493 - 503; http://dx.doi.org/10.1016/S1535-6108(02)00068-5; PMID: 12124178
- Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A 2004; 101:2452 - 7; http://dx.doi.org/10.1073/pnas.0306734101; PMID: 14983030
- Kashatus D, Cogswell P, Baldwin AS. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev 2006; 20:225 - 35; http://dx.doi.org/10.1101/gad.1352206; PMID: 16384933
- Gu L, Findley HW, Zhou M. MDM2 induces NF-kappaB/p65 expression transcriptionally through Sp1-binding sites: a novel, p53-independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia. Blood 2002; 99:3367 - 75; http://dx.doi.org/10.1182/blood.V99.9.3367; PMID: 11964305
- Ponnuswamy A, Hupp T, Fåhraeus R. Concepts in MDM2 Signaling: Allosteric Regulation and Feedback Loops. Genes Cancer 2012; 3:291 - 7; http://dx.doi.org/10.1177/1947601912454140; PMID: 23150762
- Jeong SJ, Radonovich M, Brady JN, Pise-Masison CA. HTLV-I Tax induces a novel interaction between p65/RelA and p53 that results in inhibition of p53 transcriptional activity. Blood 2004; 104:1490 - 7; http://dx.doi.org/10.1182/blood-2003-12-4174; PMID: 15155458
- Liu B, Yang R, Wong KA, Getman C, Stein N, Teitell MA, et al. Negative regulation of NF-kappaB signaling by PIAS1. Mol Cell Biol 2005; 25:1113 - 23; http://dx.doi.org/10.1128/MCB.25.3.1113-1123.2005; PMID: 15657437
- Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol 2007; 27:8284 - 95; http://dx.doi.org/10.1128/MCB.00050-07; PMID: 17908790
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J 2000; 19:94 - 102; http://dx.doi.org/10.1093/emboj/19.1.94; PMID: 10619848
- Rice NR, Ernst MK. In vivo control of NF-kappa B activation by I kappa B alpha. EMBO J 1993; 12:4685 - 95; PMID: 8223478
- Krappmann D, Scheidereit C. Regulation of NF-kappa B activity by I kappa B alpha and I kappa B beta stability. Immunobiology 1997; 198:3 - 13; http://dx.doi.org/10.1016/S0171-2985(97)80022-8; PMID: 9442373
- Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev 1993; 7:7A 1266 - 76; http://dx.doi.org/10.1101/gad.7.7a.1266; PMID: 8319912
- Lai Z, Yang T, Kim YB, Sielecki TM, Diamond MA, Strack P, et al. Differentiation of Hdm2-mediated p53 ubiquitination and Hdm2 autoubiquitination activity by small molecular weight inhibitors. Proc Natl Acad Sci U S A 2002; 99:14734 - 9; http://dx.doi.org/10.1073/pnas.212428599; PMID: 12407176
- Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005; 7:547 - 59; http://dx.doi.org/10.1016/j.ccr.2005.04.029; PMID: 15950904
- Cheng Q, Chen J. The phenotype of MDM2 auto-degradation after DNA damage is due to epitope masking by phosphorylation. Cell Cycle 2011; 10:1162 - 6; http://dx.doi.org/10.4161/cc.10.7.15249; PMID: 21386656
- Hochrainer K, Racchumi G, Zhang S, Iadecola C, Anrather J. Monoubiquitination of nuclear RelA negatively regulates NF-κB activity independent of proteasomal degradation. Cell Mol Life Sci 2012; 69:2057 - 73; http://dx.doi.org/10.1007/s00018-011-0912-2; PMID: 22261743
- Nihira K, Ando Y, Yamaguchi T, Kagami Y, Miki Y, Yoshida K. Pim-1 controls NF-kappaB signalling by stabilizing RelA/p65. Cell Death Differ 2010; 17:689 - 98; http://dx.doi.org/10.1038/cdd.2009.174; PMID: 19911008
- Gu L, Zhu N, Findley HW, Woods WG, Zhou M. Identification and characterization of the IKKalpha promoter: positive and negative regulation by ETS-1 and p53, respectively. J Biol Chem 2004; 279:52141 - 9; http://dx.doi.org/10.1074/jbc.M407915200; PMID: 15469934
- Kawauchi K, Araki K, Tobiume K, Tanaka N. Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem Biophys Res Commun 2008; 372:137 - 41; http://dx.doi.org/10.1016/j.bbrc.2008.05.021; PMID: 18477470
- Azmi AS, Banerjee S, Ali S, Wang Z, Bao B, Beck FW, et al. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget 2011; 2:378 - 92; PMID: 21623005
- Mulay SR, Thomasova D, Ryu M, Anders HJ. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int 2012; 81:1199 - 211; http://dx.doi.org/10.1038/ki.2011.482; PMID: 22297670
- Lau R, Niu MY, Pratt MA. cIAP2 represses IKKα/β-mediated activation of MDM2 to prevent p53 degradation. Cell Cycle 2012; 11:4009 - 19; http://dx.doi.org/10.4161/cc.22223; PMID: 23032264
- Criollo A, Chereau F, Malik SA, Niso-Santano M, Mariño G, Galluzzi L, et al. Autophagy is required for the activation of NFκB. Cell Cycle 2012; 11:194 - 9; http://dx.doi.org/10.4161/cc.11.1.18669; PMID: 22186785
- He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle 2011; 10:2994 - 3002; http://dx.doi.org/10.4161/cc.10.17.17118; PMID: 21862876
- Zabel U, Schreck R, Baeuerle PA. DNA binding of purified transcription factor NF-kappa B. Affinity, specificity, Zn2+ dependence, and differential half-site recognition. J Biol Chem 1991; 266:252 - 60; PMID: 1985897