Abstract
Glioblastoma (GBM) is the most aggressive primary brain tumor in human. Recent studies on high-grade pediatric GBM have identified two recurrent mutations (K27M and G34R/V) in genes encoding histone H3 (H3F3A for H3.3 and HIST1H3B for H3.1).Citation1,Citation2 The two histone H3 mutations are mutually exclusive and give rise to tumors in different brain compartments.Citation3 Recently, we4 and others5 have shown that the histone H3 K27M mutation specifically altered the di- and tri-methylation of endogenous histone H3 at Lys27. Genome-wide studies using ChIP-seq on H3.3K27M patient samples indicate a global reduction of H3K27me3 on chromatin. Remarkably, we also found a dramatic enrichment of H3K27me3 and EZH2 (the catalytic subunit H3K27 methyltransferase) at hundreds of gene loci in H3.3K27M patient cells. Here, we discuss potential mechanisms whereby H3K27me3 is enriched at chromatin loci in cells expressing the H3.3K27M mutation and report effects of Lys-to-Met mutations of other well-studied lysine residues of histone H3.1/H3.3 and H4 on the corresponding endogenous lysine methylation. We suggest that mutation(s) on histones may be found in a variety of human diseases, and the expression of mutant histones may help to address the function of histone lysine methylation and possibly other modifications in mammalian cells.
Genetic and Epigenetic Alterations in Cancers
Mutations that activate oncogenes and inactivate tumor suppressor genes are the driving forces of tumorigenesis. In the past decade, people have begun to appreciate that epigenetic change including changes in DNA methylation pattern, and posttranslational modifications on histone proteins also play critical roles in variety of pathologies.Citation6-Citation10 One typical example is the epigenetic silencing of BRCA1 in sporadic breast cancer cases.Citation11-Citation13 In addition, mutations on genes encoding histone-modifying enzymes have been identified in a number of different cancers. For example, MLL family, the histone methyltransferases responsible for the methylation of histone H3 at Lys4, is often mutated in myeloid/lymphoid or mixed-lineage leukemia cases.Citation14,Citation15 Mutations at EZH2, the catalytic subunit of the PRC2 complex responsible for the methylation of histone H3 at Lys27, were found in diffuse large B-cell lymphomaCitation16-Citation18. Recent studies using the next generation sequencing (NGS) technologies have identified for the first time mutations on histone genes.Citation1,Citation2 These two recurrent somatic mutations (K27M and G34R/V) are on histone genes encoding histone H3 variant H3.3Citation19-Citation21 in high-grade pediatric brain tumors, including diffuse intrinsic pontine glioma (DIPG) and glioblastoma multiforme (GBM).Citation22 These examples reveal the important roles of histone modifications in human diseases and potential intimate connections between genetic mutations and epigenetic changes in tumorigenesis.
Expression of H3.3K27M Mutant Results in Reduction of Endogenous H3K27me2 and H3K27me3
Lysine residues on histone H3 and H4 are often post-translationally modified to regulate chromatin structure and gene expression. For instance, mono-methylation of H3 Lys27 is enriched in gene bodies of highly expressed genes.Citation23 In contrast, di- and tri-methylation of H3 Lys27 is associated with silencing of developmental regulated genes, and the repressive histone mark “H3K27me3” also coats the inactivated X chromosome (Xi) in female mammals.Citation24-Citation26 Furthermore, H3 Lys27 can also be acetylated, and this modification is enriched at enhancers and linked to gene activation.Citation27,Citation28
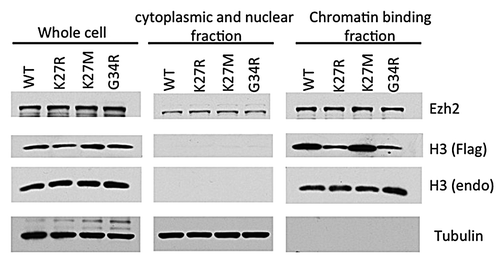
To determine whether the lysine 27 to methionine mutation found in DIPG patients affect histone modifications, we examined the levels of a variety of histone modifications by western blot. We found that the levels of di- and tri-methylation of histone H3 at Lys27 (H3K27me2 and H3K27me3) were greatly reduced in two H3.3K27M DIPG patient cell lines (SF7761and SF8628Citation29), but the methylation of other lysine residues on histone H3, such as Lys4 and Lys9, were not affected. To confirm that the reduction of methylation on Lys27 is the direct effect of K27M mutant H3, we expressed H3.1/H3.3 K27M mutant proteins in two different human cell lines and mouse embryonic fibroblast cell (MEF). We found that expression of low levels of the H3K27M transgene reduced the endogenous histone H3K27me2 and H3K27me3 in all these cell types, suggesting that the loss of H3K27me2 and H3K27me3 is due to the expression of the H3K27M transgene, and that this loss is cell type-independent.Citation4 Interestingly, we also observed the dramatic loss of H3K27me3 on the inactive X-chromosome (Xi) in female MEFs expressing the H3.1 or H3.3 K27M transgene (). Most of genes on Xi are epigenetically silenced to compensate the gene dosage between XX female and XY male mammals. H3K27me3 is not essential for the maintenance of Xi silencing. However, recruitment of PCR2 complex by XIST RNA and subsequent methylation of H3K27 is critical for the establishment of Xi silencing.Citation30-Citation33 These results demonstrate that expression of H3K27M transgene can also lead to a dramatic reduction in the levels of H3K27me3 localized at Xi, and suggest that Xi is possibly also coated with histone variant H3.3. We also found that the levels of EZH2 on chromatin were not affected in cells expressing H3K27M mutations using a chromatin fractionation assay (). Together, these results are consistent with the idea that the H3K27M mutation plays a dominant-negative role in regulating H3K27me2 and H3K27me3 levels in cells.
Figure 1. The histone H3K27M mutation reduces H3K27 methylation on Xi. Co- Immunofluorescence staining of H3K27me3 (red) and Flag (green) in iMEF cells expressed the Flag-tagged H3.3 WT or H3.3K27M histone proteins. Arrowheads indicate the Xi in iMEF expressed the Flag-tagged histones. Arrows denote Xi in un-transfected cells. Scale bar, 5 μm.
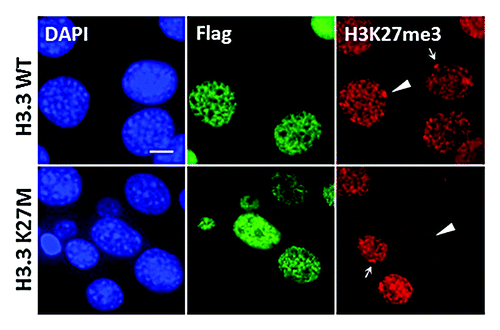
Figure 2. K27M mutants do not alter the whole EZH2 binding to the chromatin. The whole-cell lysates, cytosol, or nuclear fractions from 293T cells expressing histone wild-type (WT) H3.3, H3.3K27R, H3.3K27M, or H3.3G34R mutant transgene were prepared, and proteins in these fractions were analyzed by western blotting.
In an independent study, the Allis groupCitation5 also reported that the reduction of H3K27me2 and H3K27me3 in DIPG cancer cells as well as in cells expressing H3K27M transgene. Importantly, they show that the K27M mutant inhibits the PRC2 methyltransferase activity via interaction with the EZH2 active site. In vitro binding assays showed that EZH2, but not the other PRC2 subunits, binds to K27M peptides. Structure-functional analyses of the K27M-EZH2 reveal that the K27M histone has less inhibitory effect on a mutant EZH2 with a Tyrosine (Y) to Asparagine (N) substitution on one of the key residues in the aromatic cage of the active site.Citation5 Remarkably, H3K27I mutant is a more potent inhibitor against PCR2 than H3K27M. Together, these studies indicate that the hydrophobic interactions between the methionine side chain and the aromatic amino acid residues at the EZH2 active sites are crucial for K27M to inhibit PRC2, providing a mechanism whereby expression of H3K27M mutant results in reduced H3K27me2 and H3K27me3 levels in DIPG cells or any other cell types.
H3K27me3 Peaks Were Detected at 100 Loci in DIPG Cells
To gain additional insight into the effect of the H3.3K27M mutation, we also analyzed the localization patterns of H3K27me3, H3K4me3, and EZH2 using ChIP-seq. In general, H3K4me3 was not affected in DIPG H3.3K27M patient cells (SF7761), in reference to human neural stem cells (NSC). ChIP-seq results revealed that in addition to a global reduction of H3K27me3, “gain” of H3K27me3 in hundreds of genomic loci in SF7761 cells was observed compared with NSC control ().Citation4These gained H3K27me3-EZH2 peaks in SF7761 have several features. First, the H3K27me3 peaks unique in DIPG patient cells display a much “broader” genomic occupancy compared with normal H3K27me3 peaks found in NSCs and other cell types.Citation34 Second, these H3K27me3 “board” peaks mirror image the EZH2 localization pattern. These results strongly indicate that the gain of H3K27me3 peaks is due to the recruitment of PCR2 complex to these chromatin loci.
Figure 3. Proposed model of H3K27me3 and EZH2 peaks detected in H3.3K27M DIPG patient cells. (A) Cartoons depicting the two effects of H3K27me3 levels observed in DIPG cells. Upper panel: A global loss of H3K27me2 and H3K27me3 was observed in DIPG patient samples. The loss of repressive H3K27 methylation mark leads to reactivation of epigenetically silenced genes. Lower panel: A local gain of H3K27me3 was found in DIPG samples at hundreds of loci. Genes with the gain of H3K27me3 marks results in gene silencing. (B) Two models explaining gaining of H3K27me3 in DIPG cells. (model 1) The K27M mutant proteins have higher binding affinity to EZH2. This leads to the initial recruitment/retention of the EZH2 to the K27M nucleosomes. The initial recruitment of EZH2 will serve as a nucleation site for the spreading of PRC2 complex to a large chromatin domain. (model 2) unknown such as non-coding RNA and/or DNA sequence specific binding factor (yellow) recruit PCR2 complex and subsequent the spreading of PCR2 complex to the gene loci devoid of nucleosomes containing H3.3.
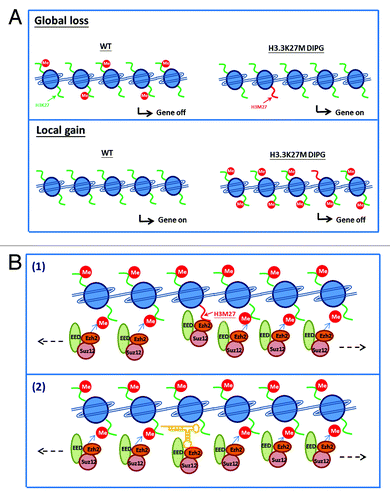
We have shown that more EZH2 binds to H3K27M containing mononucleosomes.Citation4 Peptide pull-down assays reported by the Allis groupCitation5 demonstrated the increased binding affinity of the H3K27M to EZH2. These results appear to suggest that the gain of H3K27me3 peaks is due to the recruitment of H3K27M proteins to these peaks. However, this explanation cannot account for the fact that expression of H3K27M transgene results in reduced levels of H3K27me3 globally, and H3K27M mutant proteins inhibit the activity of EZH2 in vitro. We propose 2 non-exclusive models, whereby H3K27me3 peaks are detected at hundreds loci in DIPG cells containing the H3.3K27M mutation (). First, H3K27M mutant proteins are incorporated at these loci locally, which leads to the initial recruitment of EZH2 to a define site. The initial recruitment of EZH2 will serve as a nucleation site for the spreading of PRC2 complex to a large chromatin domain. We speculate that the spreading of PRC2 complex should be independent of H3K27M mutation. In this way, PRC2 complex can be recruited to a large chromatin domain and methylate histone H3K27 in this chromatin domain. Second, the H3K27me3 peaks detected in H3.3K27M DIPG cells are chromatin domains that are devoid of H3.3 and/or H3.3K27M proteins. A specific factor including non-coding RNA and/or DNA sequence specific binding factors will recruit PCR2 complex and subsequent spreading of PCR2 complex to this domain. Further studies are needed to test these models to determine to what extent the EZH2 and H3K27me3 enriched peaks depends on the presence of H3.3K27M proteins. Whatever the mechanism is, we suspect that the H3K27me3 peaks in cells expressing H3.3K27M mutation may be cell type-specific. In addition, determination of the mechanism by which these unique H3K27me3 peaks are formed will help us understand how the H3.3K27M mutation reprograms H3K27methylation and gene expression in DIPG tumor.
Rewiring of H3K27 Methylation Is Associated with Changes in Gene Expression: Implications for the Development and Treatment of DIPG Tumors
To determine whether gain or loss of H3K27me2/me3 is associated with gene expression, we performed RNA-seq experiments. The average expression of 2975 genes (group A) that exhibit low H3K27me3 levels at their promoter in DIPG cells in reference to neural stem cells is high in DIPG cells compared with NSC, whereas the average expression of 775 genes (group C) with “gain” the H3K27me3 was significantly lower in cancer cell lines compared with NSC. Gene ontology (GO) and KEGG pathway analyses indicate that Group A genes are involved in various neurological processes, and Group C genes are involved in cancer pathways. Thus, our genome-wide ChIP-seq and gene expression analyses demonstrate that the H3.3K27M mutation rewires H3K27 methylation and gene expression. Further studies are needed to determine whether genes with increased/reduced expression or both contribute to tumorigenesis. In addition, our findings have implications for the development of a treatment strategy for patients with deadly DIPG. For instance, if genes with the “gain” of H3K27me3 observed in cells expressing H3K27M mutation is the primary driver to tumorigenesis, one would expect that cancer cells treated with inhibitors against EZH2Citation35,Citation36 should specifically target cells expressing H3K27M mutant. Alternatively, if genes with the loss of H3K27methylation are the primary driver for tumorigenesis, one would expect that inhibitors against H3K27 demethylases should help treating this deadly disease.Citation37-Citation40 Finally, because of the reprogramming of H3K27 methylation and possibly other epigenetic marks in H3.3K27M cancer cells, we hypothesize that these cancer cells will be addictive to other epigenetic regulators for proliferation and migration. Therefore, discovery of these epigenetic regulators will also hold potential for the treatment of pediatric GBM as well. We expect that future studies aimed at tackling these questions/possibilities will yield information and treatment strategy of this deadly disease.
Effect of Expressing K-to-M Mutations of Other Lysine Residues on Histone H3 and H4 Methylation
Expression of the H3K27M mutant transgene results in reduced levels of H3K27me3 on endogenous histone H3 histone.Citation4,Citation5 We determined whether the same substitution on other lysine residues would also specifically affect methylation of the corresponding lysine residues of endogenous histones. To address this, we mutated 5 well-studied lysine residues on both histone H3.1 and H3.3 (Lys4, 9, 27, 36, 79) and the Lys20 on histone H4 to methionine (), transiently expressed each of the transgenes in 293T, and analyzed the effect on histone methylation (). Expression of a transgene containing Lys-to-Met substitution of lysine 27 and Lys36 reduced the levels of the methylation on the corresponding residues of endogenous histone H3 dramatically. The Allis groupCitation5 has also reported that expression of H3K9M and H3K36M transgenes also lead to reduction in methylation of corresponding endogenous lysine residues. In contrast, Lys to Met mutation of H3K4, H3K79, and H4K20 had no obvious effect on the methylation of endogenous histone in cells transiently expressing each histone mutant (). We have previously shown that it takes multiple generations for the H3K27M mutant to exert its effect on H3K27me3 in cells.Citation4 Therefore, we tested whether the endogenous methylation is affected in cells stably expressing the H3K4M, H3K79M, and H4K20M transgene. For each mutant, we have examined the levels of histone methylation in at least 2 independent cell lines. In all lines we have tested, we found that the endogenous methylation of H3K4, H3K9, H3K79, and H4K20 were markedly reduced in 293T stably expressing the corresponding Lys-to-Met mutant (). These data are consistent with the idea that different Lys-Met mutants may have different inhibition efficiencies on their corresponding KMTs in vivo.
Figure 4. Graphical summary of the lysine residues on histone H3.1/H3.3/H4 that were mutated and tested in this study. Lys4, 9, 27, 36 of histone H3.1 and H3.3 and Lys20 of histone H4 are located at the N-terminal tail. Lys79 of histone H3 is located in the histone core. Some of the lysine methyltransferases are listed in the boxes.
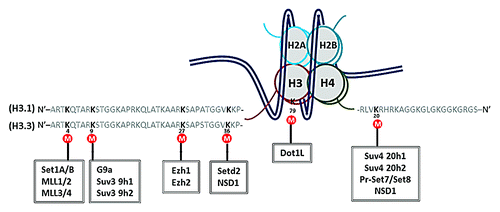
Figure 5. The effect of Lys-to-Met mutation on the methylation of the corresponding lysine residue of endogenous histones (A) Whole-cell lysates from 293T cells transiently expressing the “K to M” mutant histone proteins were collected and subjected to western blot. Histone modifications were detected with the indicated antibodies. (B–E) Whole-cell lysates from 293T cells stably expressing the indicated mutant histones were collected and subjected to western blot. Two independent lines were analyzed for the effect of each mutant on histone methylation.
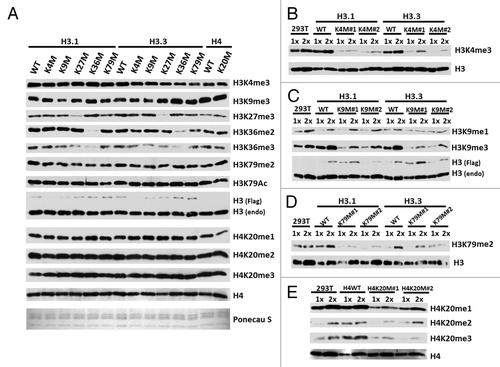
Unlike other commonly modified lysine residues located at the N-terminal tail, H3K79 locates within the histone globular domain (). In addition, methylation at H3K79 is catalyzed by DOT1/DOT1L, which does not have the classical SET domain present in other lysine methyltransferases.Citation41,Citation42 Our results suggest the possibility that all the K-M mutants can inhibit the corresponding HMT(s) activities, regardless of the location of the Lys residues on histones and/or the catalytic domains used for catalysis.
Perspective
H3.3K27M mutation in DIPG cells reprograms H3K27 methylation on endogenous wild-type histone. We have shown here other Lys-Met substitutions on histone H3 and H4 also affect the endogenous methylation () and possibly alter methylation pattern of the corresponding lysine residues and gene expression. These findings raise a number of questions. First, is it possible to detect mutation at other histone lysine residues in other tumors and/or diseases? Second, in addition to lysine methylation, can the substitution of arginine, serine, and lysine residues on histones with a specific amino acid affect arginine methylation,Citation43-Citation46 serine phosphorylation,Citation47-Citation49 and lysine ubiquitinationCitation50,Citation51 of endogenous histones? Finally, genome-wide studies indicate that a histone modification generally has a unique localization pattern on chromatin;Citation52-Citation54 however, it is unknown whether the unique localization profile of a particular modification has any functional implications. In yeast, it is possible to mutate a specific histone residue of interest and study the function of individual histone modification.Citation55-Citation61 Recently, this method has extended to study the function of histone modification in Drosophila.Citation62 However, this approach is not applicable in mammalian cells because of the large number of genes encoding the histone proteins.Citation63 For this reason, almost all the studies on the function of histone modification in mammalian cells utilize siRNA knockdown or small-molecule inhibition of the histone-modifying enzymes. These approaches have obvious drawbacks, such as off-target effects of siRNACitation64 and inhibitors. In addition, most of histone-modifying enzymes have non-histone substrates.Citation65 For example, EZH2 generates a methyl degron on non-histone proteins. These make the analysis of the cellular phenotypes difficult and possibly generate false positive/negative results. Finally, mutations at histones in yeast and Drosophila and inhibition of enzymes using small-molecule inhibitors only reduce the level, but not alter the pattern, of a particular modification. Is it possible to use the Lys-to-Met mutation on histone lysine methylation described here and by Lewis et al.Citation5 to specifically change the modification pattern of a particular lysine methylation in cells? Do alterations in H3K27me3, H3K4me3, or a particular epigenetic landscape in stem cells affect stem cell differentiation and maintenance? Do changes in patterns in H3K36me3 and H4K20me3, 2 histone marks that have been documented to be important for DNA response and repair, affect DNA repair?Citation66,Citation67 We expect that studies aimed at addressing these questions will significantly enrich our understanding of the potential functions of histone modifications in different cellular processes and in disease conditions where alterations in histone modifications have been linked.
Methods and Materials
Cell culture
Female immortalized MEF cells have been described.Citation68 293T cells were cultured under standard conditions.
Plasmids
Full-length and mutated forms of human histone H3.1 and H3.3 cDNA were cloned into pQCXIP vector for expression in 293T cells and iMEF.
Antibodies
H3K27me2 (#9728), H3K27me3 (#9733), EZH2 (#5246), H3K36me2 (#2901). H3K36me3 (#4909) were purchased from Cell Signaling. H3K4me3 (#07-473) were purchased from Millipore. α-Tubulin (#T9026) and Flag (F1804) antibodies were purchased from Sigma. H3K79me2 (ab3594) was purchased from Abcam. H3K9me1, H3K9me3, H3K79Ac, H4K20me1, H4K20me2, H4K20me3, and histone H3 antibodies have been described.Citation69
Preparation of chromatin and cytosol fractions
Cells were harvested in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, 1 mM DTT) and then incubated on ice for 5 min. Cytoplasmic fraction and nuclei were separated by centrifugation at 1300 g, 4 °C, 4 min. The nuclear pellet was washed once with buffer A and resuspended in Laemmli buffer to serve as the chromatin fraction. The supernatant was further clarified by centrifugation at 20 000 g, 4 °C, 15 min and was treated as the cytoplasmic fraction.
Acknowledgments
KMC is supported by Mayo Edward C Kendall Fellowship. These studies are supported by grants from the National Institutes of Health (CA157489 and GM99722) and by Epigenomic Developmental Program at the Center of Individualized Medicine, Mayo Clinic. ZZ is a Scholar of Leukemia and Lymphoma Society.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482:226 - 31; http://dx.doi.org/10.1038/nature10833; PMID: 22286061
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al, St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012; 44:251 - 3; http://dx.doi.org/10.1038/ng.1102; PMID: 22286216
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22:425 - 37; http://dx.doi.org/10.1016/j.ccr.2012.08.024; PMID: 23079654
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27:985 - 90; http://dx.doi.org/10.1101/gad.217778.113; PMID: 23603901
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340:857 - 61; http://dx.doi.org/10.1126/science.1232245; PMID: 23539183
- Elsässer SJ, Allis CD, Lewis PW. Cancer. New epigenetic drivers of cancers. Science 2011; 331:1145 - 6; http://dx.doi.org/10.1126/science.1203280; PMID: 21385704
- You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin?. Cancer Cell 2012; 22:9 - 20; http://dx.doi.org/10.1016/j.ccr.2012.06.008; PMID: 22789535
- Swami M. Epigenetics: Demethylation links cell fate and cancer. Nat Rev Genet 2010; 11:749; http://dx.doi.org/10.1038/nrg2890; PMID: 20921960
- Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med 2011; 17:330 - 9; http://dx.doi.org/10.1038/nm.2305; PMID: 21386836
- Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev 2004; 18:2315 - 35; http://dx.doi.org/10.1101/gad.1232504; PMID: 15466484
- Rice JC, Ozcelik H, Maxeiner P, Andrulis I, Futscher BW. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis 2000; 21:1761 - 5; http://dx.doi.org/10.1093/carcin/21.9.1761; PMID: 10964110
- Rice JC, Futscher BW. Transcriptional repression of BRCA1 by aberrant cytosine methylation, histone hypoacetylation and chromatin condensation of the BRCA1 promoter. Nucleic Acids Res 2000; 28:3233 - 9; http://dx.doi.org/10.1093/nar/28.17.3233; PMID: 10954590
- Tapia T, Smalley SV, Kohen P, Muñoz A, Solis LM, Corvalan A, et al. Promoter hypermethylation of BRCA1 correlates with absence of expression in hereditary breast cancer tumors. Epigenetics 2008; 3:157 - 63; http://dx.doi.org/10.4161/epi.3.3.6387; PMID: 18567944
- Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev 2011; 25:661 - 72; http://dx.doi.org/10.1101/gad.2015411; PMID: 21460034
- Mohan M, Lin C, Guest E, Shilatifard A. Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer 2010; 10:721 - 8; http://dx.doi.org/10.1038/nrc2915; PMID: 20844554
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012; 109:2989 - 94; http://dx.doi.org/10.1073/pnas.1116418109; PMID: 22323599
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010; 42:181 - 5; http://dx.doi.org/10.1038/ng.518; PMID: 20081860
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013; 23:677 - 92; http://dx.doi.org/10.1016/j.ccr.2013.04.011; PMID: 23680150
- Ahmad K, Henikoff S. Histone H3 variants specify modes of chromatin assembly. Proc Natl Acad Sci U S A 2002; 99:Suppl 4 16477 - 84; http://dx.doi.org/10.1073/pnas.172403699; PMID: 12177448
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 2002; 9:1191 - 200; http://dx.doi.org/10.1016/S1097-2765(02)00542-7; PMID: 12086617
- Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res 2011; 21:421 - 34; http://dx.doi.org/10.1038/cr.2011.14; PMID: 21263457
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 2012; 124:439 - 47; http://dx.doi.org/10.1007/s00401-012-0998-0; PMID: 22661320
- Steiner LA, Schulz VP, Maksimova Y, Wong C, Gallagher PG. Patterns of histone H3 lysine 27 monomethylation and erythroid cell type-specific gene expression. J Biol Chem 2011; 286:39457 - 65; http://dx.doi.org/10.1074/jbc.M111.243006; PMID: 21937433
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003; 300:131 - 5; http://dx.doi.org/10.1126/science.1084274; PMID: 12649488
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002; 298:1039 - 43; http://dx.doi.org/10.1126/science.1076997; PMID: 12351676
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011; 469:343 - 9; http://dx.doi.org/10.1038/nature09784; PMID: 21248841
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010; 107:21931 - 6; http://dx.doi.org/10.1073/pnas.1016071107; PMID: 21106759
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108 - 12; http://dx.doi.org/10.1038/nature07829; PMID: 19295514
- Hashizume R, Smirnov I, Liu S, Phillips JJ, Hyer J, McKnight TR, et al. Characterization of a diffuse intrinsic pontine glioma cell line: implications for future investigations and treatment. J Neurooncol 2012; 110:305 - 13; http://dx.doi.org/10.1007/s11060-012-0973-6; PMID: 22983601
- Panning B, Dausman J, Jaenisch R. X chromosome inactivation is mediated by Xist RNA stabilization. Cell 1997; 90:907 - 16; http://dx.doi.org/10.1016/S0092-8674(00)80355-4; PMID: 9298902
- Lee JT, Jaenisch R. The (epi)genetic control of mammalian X-chromosome inactivation. Curr Opin Genet Dev 1997; 7:274 - 80; http://dx.doi.org/10.1016/S0959-437X(97)80138-4; PMID: 9115428
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961; 190:372 - 3; http://dx.doi.org/10.1038/190372a0; PMID: 13764598
- Chow J, Heard E. X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol 2009; 21:359 - 66; http://dx.doi.org/10.1016/j.ceb.2009.04.012; PMID: 19477626
- Kidder BL, Hu G, Zhao K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat Immunol 2011; 12:918 - 22; http://dx.doi.org/10.1038/ni.2117; PMID: 21934668
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492:108 - 12; http://dx.doi.org/10.1038/nature11606; PMID: 23051747
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A 2012; 109:21360 - 5; http://dx.doi.org/10.1073/pnas.1210371110; PMID: 23236167
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007; 449:731 - 4; http://dx.doi.org/10.1038/nature06145; PMID: 17713478
- Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A 2007; 104:18439 - 44; http://dx.doi.org/10.1073/pnas.0707292104; PMID: 18003914
- Miller SA, Mohn SE, Weinmann AS. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell 2010; 40:594 - 605; http://dx.doi.org/10.1016/j.molcel.2010.10.028; PMID: 21095589
- Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012; 488:404 - 8; http://dx.doi.org/10.1038/nature11262; PMID: 22842901
- Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev 2011; 25:1345 - 58; http://dx.doi.org/10.1101/gad.2057811; PMID: 21724828
- Frederiks F, Tzouros M, Oudgenoeg G, van Welsem T, Fornerod M, Krijgsveld J, et al. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol 2008; 15:550 - 7; http://dx.doi.org/10.1038/nsmb.1432; PMID: 18511943
- Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Front Biosci 2006; 11:344 - 55; http://dx.doi.org/10.2741/1802; PMID: 16146736
- Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004; 306:279 - 83; http://dx.doi.org/10.1126/science.1101400; PMID: 15345777
- Torres-Padilla ME, Parfitt DE, Kouzarides T, Zernicka-Goetz M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 2007; 445:214 - 8; http://dx.doi.org/10.1038/nature05458; PMID: 17215844
- Kirmizis A, Santos-Rosa H, Penkett CJ, Singer MA, Vermeulen M, Mann M, et al. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 2007; 449:928 - 32; http://dx.doi.org/10.1038/nature06160; PMID: 17898715
- Zippo A, De Robertis A, Serafini R, Oliviero S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol 2007; 9:932 - 44; http://dx.doi.org/10.1038/ncb1618; PMID: 17643117
- Lau AT, Lee SY, Xu YM, Zheng D, Cho YY, Zhu F, et al. Phosphorylation of histone H2B serine 32 is linked to cell transformation. J Biol Chem 2011; 286:26628 - 37; http://dx.doi.org/10.1074/jbc.M110.215590; PMID: 21646345
- Kang B, Pu M, Hu G, Wen W, Dong Z, Zhao K, et al. Phosphorylation of H4 Ser 47 promotes HIRA-mediated nucleosome assembly. Genes Dev 2011; 25:1359 - 64; http://dx.doi.org/10.1101/gad.2055511; PMID: 21724829
- Zhang Y. Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev 2003; 17:2733 - 40; http://dx.doi.org/10.1101/gad.1156403; PMID: 14630937
- Zhang F, Yu X. WAC, a functional partner of RNF20/40, regulates histone H2B ubiquitination and gene transcription. Mol Cell 2011; 41:384 - 97; http://dx.doi.org/10.1016/j.molcel.2011.01.024; PMID: 21329877
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 26; http://dx.doi.org/10.1016/j.cell.2006.02.041; PMID: 16630819
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al, ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57 - 74; http://dx.doi.org/10.1038/nature11247; PMID: 22955616
- Burgess RJ, Zhou H, Han J, Zhang Z. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell 2010; 37:469 - 80; http://dx.doi.org/10.1016/j.molcel.2010.01.020; PMID: 20188666
- Li Q, Fazly AM, Zhou H, Huang S, Zhang Z, Stillman B. The elongator complex interacts with PCNA and modulates transcriptional silencing and sensitivity to DNA damage agents. PLoS Genet 2009; 5:e1000684; http://dx.doi.org/10.1371/journal.pgen.1000684; PMID: 19834596
- Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, et al. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 2008; 134:244 - 55; http://dx.doi.org/10.1016/j.cell.2008.06.018; PMID: 18662540
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007; 315:653 - 5; http://dx.doi.org/10.1126/science.1133234; PMID: 17272723
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009; 459:802 - 7; http://dx.doi.org/10.1038/nature08085; PMID: 19516333
- Nakanishi S, Sanderson BW, Delventhal KM, Bradford WD, Staehling-Hampton K, Shilatifard A. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat Struct Mol Biol 2008; 15:881 - 8; http://dx.doi.org/10.1038/nsmb.1454; PMID: 18622391
- Yuan J, Pu M, Zhang Z, Lou Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 2009; 8:1747 - 53; http://dx.doi.org/10.4161/cc.8.11.8620; PMID: 19411844
- Pengelly AR, Copur O, Jäckle H, Herzig A, Müller J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 2013; 339:698 - 9; http://dx.doi.org/10.1126/science.1231382; PMID: 23393264
- Henikoff S, Ahmad K. Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol 2005; 21:133 - 53; http://dx.doi.org/10.1146/annurev.cellbio.21.012704.133518; PMID: 16212490
- Ui-Tei K, Naito Y, Nishi K, Juni A, Saigo K. Thermodynamic stability and Watson-Crick base pairing in the seed duplex are major determinants of the efficiency of the siRNA-based off-target effect. Nucleic Acids Res 2008; 36:7100 - 9; http://dx.doi.org/10.1093/nar/gkn902; PMID: 18988625
- Lee JM, Lee JS, Kim H, Kim K, Park H, Kim JY, et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell 2012; 48:572 - 86; http://dx.doi.org/10.1016/j.molcel.2012.09.004; PMID: 23063525
- Li F, Mao G, Tong D, Huang J, Gu L, Yang W, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 2013; 153:590 - 600; http://dx.doi.org/10.1016/j.cell.2013.03.025; PMID: 23622243
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006; 127:1361 - 73; http://dx.doi.org/10.1016/j.cell.2006.10.043; PMID: 17190600
- Chan KM, Zhang H, Malureanu L, van Deursen J, Zhang Z. Diverse factors are involved in maintaining X chromosome inactivation. Proc Natl Acad Sci U S A 2011; 108:16699 - 704; http://dx.doi.org/10.1073/pnas.1107616108; PMID: 21940502
- Chan KM, Zhang Z. Leucine-rich repeat and WD repeat-containing protein 1 is recruited to pericentric heterochromatin by trimethylated lysine 9 of histone H3 and maintains heterochromatin silencing. J Biol Chem 2012; 287:15024 - 33; http://dx.doi.org/10.1074/jbc.M111.337980; PMID: 22427655