Abstract
ERCC1 (excision repair cross-complementation group 1) plays essential roles in the removal of DNA intrastrand crosslinks by nucleotide excision repair, and that of DNA interstrand crosslinks by the Fanconi anemia (FA) pathway and homology-directed repair processes (HDR). The function of ERCC1 thus impacts on the DNA damage response (DDR), particularly in anticancer therapy when DNA damaging agents are employed. ERCC1 expression has been proposed as a predictive biomarker of the response to platinum-based therapy. However, the assessment of ERCC1 expression in clinical samples is complicated by the existence of 4 functionally distinct protein isoforms, which differently impact on DDR. Here, we explored the functional competence of each ERCC1 protein isoform and obtained evidence that the 202 isoform is the sole one endowed with ERCC1 activity in DNA repair pathways. The ERCC1 isoform 202 interacts with RPA, XPA, and XPF, and XPF stability requires expression of the ERCC1 202 isoform (but none of the 3 others). ERCC1-deficient non-small cell lung cancer cells show abnormal mitosis, a phenotype reminiscent of the FA phenotype that can be rescued by isoform 202 only. Finally, we could not observe any dominant-negative interaction between ERCC1 isoforms. These data suggest that the selective assessment of the ERCC1 isoform 202 in clinical samples should accurately reflect the DDR-related activity of the gene and hence constitute a useful biomarker for customizing anticancer therapies.
Introduction
The structure specific endonuclease activity of the XPF (xeroderma pigmentosum complementation group F)/ERCC1 (excision repair cross-complementation group 1) complex is required for the repair of several types of DNA lesions, including intra- and interstrand crosslinks as well as DNA double-strand breaks. A key role of the ERCC1/XPF heterodimer was first demonstrated in the nucleotide excision repair (NER) pathway. The 5′–3′ structure-specific endonuclease activity of the complex was subsequently shown to be also required for the Fanconi anemia (FA) pathway of interstrand crosslink repair (ICL-R),Citation1-Citation3 as well as for homology-directed repair (HDR) of DNA double-strand breaks (DSB).Citation4-Citation7 XPF is catalytically active, while ERCC1 tethers the activity of the complex to the site of repair through physical interactions with the core machinery complexes of several repair pathways (see ref. Citation8 for review). During NER, ERCC1 interacts with XPA (xeroderma pigmentosum group A) at the pre-incision complex.Citation9 Interaction of both XPF and ERCC1 with RPA (replication protein A) is then triggered and enables the optimal positioning of the complex.Citation10-Citation13 Fewer molecular details are known about the role of the XPF/ERCC1 complex in ICL-R. The interaction of the complex with FANCG (Fanconi anemia complementation group G) may underlie the role for ERCC1 in the FA pathway. Similarly, the physical and functional interaction of XPF/ERCC1 with SLX4 (a.k.a. BTBD12, BTB domain-containing protein 12, or Fanconi anemia complementation group P, FANCP) mediates the role of the heterodimer in the resolution of specific DNA structures formed during the HDR of DSB, ICL, or collapsed replication forks.Citation3,Citation6,Citation14,Citation15,Citation16
The disruption of ERCC1 leads to mitotic defects generating polyploidy and multinucleation through a mechanism that remains to be determined.Citation17,Citation18 Interestingly, a similar phenotype has been described in FA cells, raising the possibility that the impact of ERCC1 disruption on mitosis is related to its role in FA pathway.Citation17
Given the central role of ERCC1 in multiple DNA repair pathways, ERCC1 expression has been thoroughly examined in cancer samples for its potential clinical relevance, in particular in therapeutic contexts where DNA repair capacity may determine treatment outcome. ERCC1 has indeed been proposed as a prognostic and predictive biomarker in NSCLC as well as in multiple other tumor types.Citation19 So far, the implementation of ERCC1 status determination into clinical practice has been hampered by the technical challenge of assessing its activity in tumor samples. Notably, the expression of ERCC1 as 4 functionally distinct splice variants (namely isoforms 201, 202, 203, and 204) is an obvious obstacle to the accurate evaluation of ERCC1 activity [Ensemble Genome Browser], as current approaches using immunohistochemistry (IHC) or RT-PCR (reverse transcription polymerase chain reaction) cannot discriminate between them. Isoform 202, which is generally the most highly expressed one in tissue and cellular models, has been widely studied, whereas the function and activity of other isoforms are still largely unknown. Notably, ERCC1 isoform 202 reportedly is the only active isoform with regards to the removal of platinum adducts.Citation20 It is therefore crucial to investigate the contribution of each individual ERCC1 isoform to each of the DNA repair pathways relevant to the response to DNA-damaging agents.
In this report we examined the contribution of each ERCC1 isoform to the DNA repair pathways involved in the processing of cisplatin-induced DNA damages by using recently described isogenic NSCLC models of ERCC1 deficiency.Citation20 Through the exploration of the ability of the different isoforms to interact with partners relevant for NER and ICL-R pathways, we show that isoform 202 is the only functional ERCC1 product. Functional assays revealed that isoform 202 is uniquely able to rescue the polyploid and multinucleated phenotype associated with ERCC1 loss in NSCLC cell lines. We finally examined the potential dominant-negative activity of the other isoforms but found no evidence for such regulating function.
Results
Subcellular localization of ERCC1 isoforms
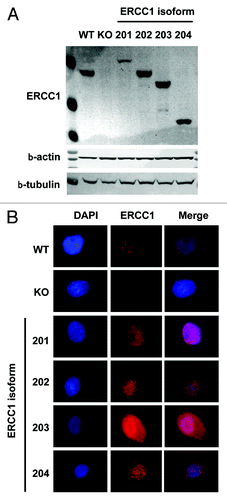
NSCLC-derived A549 cells were knocked out for ERCC1 using the Zn finger nuclease technology and then transduced with lentiviral vectors driving the ectopic re-expression of each of the 4 ERCC1 isoforms (). To gain insight into the activity of the different ERCC1 isoforms, we first examined the subcellular localization of each isoform by immunofluorescence microscopy using the FL297 anti-ERCC1 antibody, a polyclonal rabbit antibody that detects all 4 isoforms. As expected, we only detected background signals in the ERCC1-deficient cells, compared with the parental wild type (WT) or the single-isoform re-expressing clones (). In WT cells, ERCC1 was detected in the nucleus and could form nuclear foci, possibly reflecting repair processes of basal DNA damages. Each of the 4 isoforms also localized in the nucleus, with nuclear foci observed in the single-isoform expressing clones. Interestingly, isoform 203 additionally generated a significant cytoplasmic signal. Therefore, the 4 ERCC1 isoforms showed expression patterns that were comparable to the endogenous ERCC1 products and compatible with the known ERCC1 biological activity.
Figure 1. Subcellular localization of ERCC1 isoforms. (A) ERCC1 expression was assessed by immunoblotting in wild-type A549 (WT), A549 knocked-down for ERCC1 (KO) and A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204). β-actin and β-tubulin were used as loading controls. (B) Immunofluorescence staining of wt-A549 (WT), A549 knocked-down for ERCC1 (KO) and A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204) for ERCC1 (FL297 antibody, red). Nuclear DNA was counterstained with DAPI (blue).
ERCC1 isoform 202 interacts with partners involved in NER
The best-characterized role of ERCC1 involves its heterodimerization with the enzymatically active XPF protein. The heterodimer possesses a structure-specific nuclease activity and catalyzes crucial biochemical reactions required for the repair of bulky DNA adducts. We have previously provided evidence for the strict requirement of the ERCC1 isoform 202 to the processing of cisplatin adducts and subsequent cell survival.Citation20 In order to gain mechanistic understanding of the unique activity of the 202 isoform, we sought to examine the ability of each ERCC1 isoform to interact with partners essential for its function in the NER pathway.Citation21 Using the proximity ligation assays (PLA, Duolink®) technology, we first examined the ability of the ERCC1 isoforms to interact with XPF (). As expected, the punctuated signal observed in WT A549 cells indicated that ERCC1 and XPF were interacting at sites of DNA repair. No signal was detected in the ERCC1-deficient cell lines, except for the ERCC1–202 re-expressing line. These data strongly suggest that the ERCC1 isoform 202 is the only gene product able to physically interact with XPF and co-localize at nuclear foci.
Figure 2. ERCC1 interaction with NER factors. (A) Representative images of proximity ligation assay (PLA, Duolink®) detection of ERCC1/XPF heterodimers (red) in wild-type A549 (WT), A549 knocked-down for ERCC1 (KO) and A549 expressing individually each of the 4 ERCC1 isoform (201, 202, 203, and 204). Nuclear DNA was counterstained with DAPI (blue). (B) Quantification of PLA assay detection of the interaction between ERCC1 and RPA, XPA, and XPF performed as in (A). The nuclear dots per nucleus were counted using the ImageJ software. The ratio of the signal in KO and single isoform-expressing A549 over WT cells was plotted.
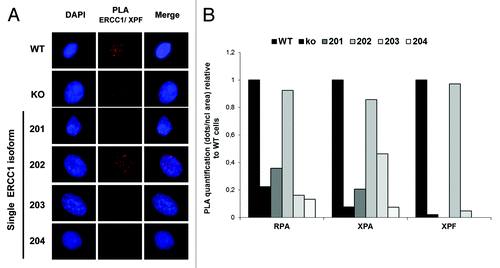
The enzymatically active ERCC1/XPF heterodimer interacts with additional NER factors such as XPA and RPA, which contribute to its proper localization and activity during DNA repair.Citation9,Citation10-Citation13,Citation21 We therefore also explored the ability of the ERCC1 isoforms to interact with these factors in PLA (). While the PLA signal was reduced in ERCC1-deficient cells, only ERCC1 isoform 202 could restore signal levels comparable to WT cells, highlighting its unique ability to interact with RPA, XPA, and XPF and to form active NER complexes.
ERCC1–202 is required for XPF expression
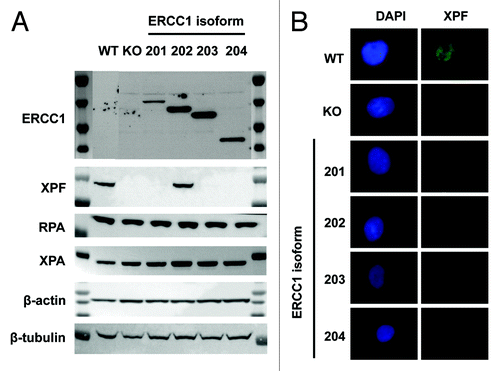
Previous studies have suggested that XPF is unstable in the absence of interaction with ERCC1.Citation22 As ERCC1–202 was the only isoform able to interact with XPF in PLA, we then tested whether an isoform-specific impact on XPF protein level could be observed. XPF, XPA, and RPA protein expression levels were examined by western blot in each of the ERCC1-deficient cells lines expressing a single ERCC1 isoform. While no impact of the ERCC1 loss was observed on XPA and RPA protein levels, XPF protein levels showed a clear dependency on the presence of the ERCC1 isoform 202 (). We confirmed by immunofluorescence microscopy that the expression of the ERCC1–202 was required for an XPF signal to be detected in the nucleus (). These results strongly support that ERCC1 isoform 202 is an obligate interacting partner of XPF.
Figure 3. ERCC1–202 is required for XPF expression. (A) Immunoblot analysis of ERCC1, XPF, RPA, and XPA in wild-type A549 (WT), A549 knocked-down for ERCC1 (KO), and A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204). β-actin and β-tubulin were used as loading controls. (B) Immunofluorescence staining of wt-A549 (WT), A549 knocked-down for ERCC1 (KO), and A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204) for XPF (FL297 antibody, red), as in . Nuclear DNA was counterstained with DAPI (blue). Scale bar, 10 µm.
ERCC1 deficiency leads to multinucleation
ERCC1 knockdown in mouse and human cells leads to nuclear and cytoplasmic morphological alterations that may result from abnormal mitosis.Citation17,Citation18,Citation23-Citation25 We therefore initially sought to investigate whether the phenotype of NSCLC cells would be altered by ERCC1 knockout. Cytological staining of ERCC1-deficient cells indeed confirmed the presence of morphological alterations, including enlarged nuclei and cytoplasms, as well as cases of multinucleation, which were not found in ERCC1-proficient control cells (). The quantitative assessment of the mean nuclear size and of the percentage of multinucleated cells showed a consistent increase in ERCC1-deficient cells ( and , 6- and 2-fold, respectively). As expected from these observations, the ploidy of the ERCC1-deficient cells was increased accordingly when assessed by flow cytometric DNA profiling (, 0.08% of 4N in WT cells vs. 1.13% in ERCC1-deficient clones).
Figure 4. ERCC1 deficiency-induced polyploidy in NSCLC cells is rescued by the 202 isoform. (A) Diff-QuickTM staining of wild-type A549 (WT), A549 knocked-down for ERCC1 (KO), and A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204). Arrowheads point to abnormally sized nuclei and multinucleated cells. Scale bar, 20 µm. (B) Nuclear areas were determined using ImageJ software in wild-type A549 (WT), A549 knocked-down for ERCC1 (KO), and A549 expressing individually each of the 4 ERCC1 isoform (201, 202, 203, and 204) stained with Diff-Quick™ as in (A). The percentage of cells with nuclear area superior to the average nuclear area of WT cells was plotted. Error bars indicate SEM. (C) The percentage of multinucleated cells was scored manually on Diff Quick™ stain cells (n = 200). Error bars indicate SEM. (D) wild-type A549 (WT), A549 knocked-down for ERCC1 (KO) and A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204) were blocked in G2/M by a 6 h colcemid treatment (Karyomax) and then processed for PI DNA profiling. Dot plot representation of the flow cytometry analysis is shown. Circled population corresponds to aneuploidy single cells containing more than 4N DNA. (E) Images show representative γ- and α-tubulin immunofluorescence staining of A549 cells used to score for centrosome number per cell (scale bar, 10 µm). The percentage of cells with more than 2 centrosomes was plotted. At least 200 wild-type A549 (WT), A549 knocked-down for ERCC1 (KO), or A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204) were counted. Error bars indicate SEM.
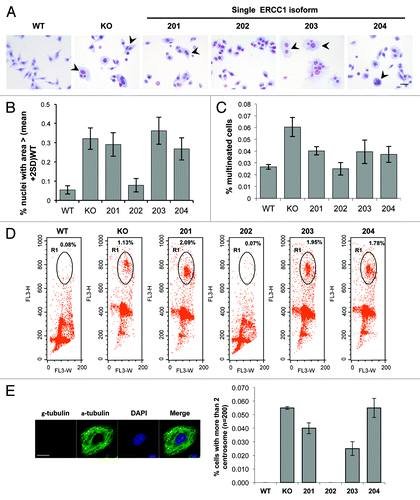
Polyploidy can arise from cytokinesis failure or cell fusion, both leading to concomitant centrosome amplification. When examined by γ-tubulin immunofluorecence staining, the number of centrosome per cells was indeed increased significantly in the ERCC1-deficient cells (). Taken together, these data suggest that the loss of ERCC1 in NSCLC cells generates polyploid multinucleated cells.
ERCC1–202 isoform rescues the ERCC1-deficiency induced multinucleation
The capacity of each isoform to reverse the aforementioned cytotological phenotype of ERCC1-deficient cells was then addressed. Strikingly, all of the alterations observed in ERCC1-deficient cells could be rescued exclusively by the re-expression of the 202 isoform. Instead, the re-expression of the 201, 203, and 204 isoform did not impact on morphological alterations driven by ERCC1 loss, including the increased ploidy and centrosome numbers. On the contrary, ERCC1-deficient cells ectopically expressing the 202 isoform displayed a similar phenotype as WT cells ().
The presence of anaphase bridges in ERCC1-deficient cells is associated with cytokinesis failure
The mechanistic basis for the polyploid phenotype of ERCC1-deficient cells is still not firmly established and may arise either from cell fusion or cytokinesis failure. shows representative snapshots from time-lapse fluorescence microscopy of mitotic ERCC1-deficient cells displaying cytoplasmic bridges and failing to complete cytoplasmic abscission. The 2 daughter cells that are on the verge of separating from each other eventually fuse back together, pointing to cytokinesis failure as a likely mechanism for the generation of polyploid cells by the ERCC1 disruption.
Figure 5. The role of ERCC1 in interstrand cross-links repair is fulfilled by the 202 isoform. (A) Time-lapse video microscopy was performed on ERCC1-deficient A549 cells maintained in growing medium containing tracker green and Hoechst 33342 used for cytoplasmic and nuclear staining, respectively. Representative snapshots of 4 failed mitoses are shown. Scale bar, 20 µm. (B) Immunofluorescence images of ERCC1-deficient A549 cells stained for α-tubulin for visualization of mitotic spindles and counterstained with DAPI. DNA bridges are visible in anaphase as well as during cytokinesis. Scale bar, 10 µm. (C) ERCC1 interaction with the FA gene products FANCG and SLX4 was assessed in PLA. The PLA signal detected in wild-type A549 (WT), A549 knocked-down for ERCC1 (KO), and A549 expressing individually each of the 4 ERCC1 isoforms (201, 202, 203, and 204) was quantified using the ImageJ software. The ratio of the signal in KO and single-isoform-expressing A549 over WT cells was plotted. (D) Wild-type A549 (WT), A549 knocked-down for ERCC1 (KO), and A549 expressing individually each of the four ERCC1 isoform (201, 202, 203, and 204) were treated for 48 h with Mitomycin C or with vehicle as indicated. Cell viability was then assessed by WST-1 assay. Percentage of surviving cells was plotted and IC50 determined. Error bars indicate the SEM of triplicate measurements of a representative experiment.
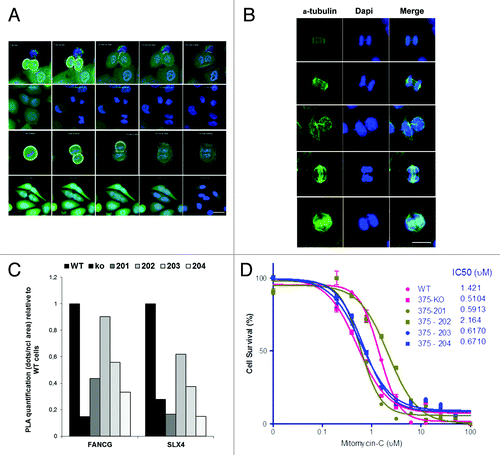
A common cause for cytokinesis failure is the presence of intertwined sister chromatids that can form DNA bridges during mitosis and prevent cytoplasm abscission. The presence of anaphase bridges in ERCC1-deficient cells was revealed by immunofluorescence microscopy (), thus providing a potential mechanistic explanation for the mitotic defects observed in ERCC1-deficient cells.
The ERCC1–202 isoform participates to the processing of anaphase bridge-generating DNA structures
The persistence of incompletely processed DNA lesions arising during S or G2 phase from stalled replication forks and the presence of ICLs or DSB represents a potential source of anaphase bridges that can result in cytokinesis failure.Citation26 The repair of these lesions relies on the FA pathway and on homology-directed repair, 2 processes likely to be affected by ERCC1 disruption.Citation27,Citation28 Indeed, ERCC1 participates to the FA-mediated repair of ICLs and was shown to interact with FANCG.Citation3,Citation14 Moreover the structure-specific endonuclease activity of XPF/ERCC1 is also required for the completion of HDR, and this function of the complex involves its interaction with the FA pathway gene SLX4.Citation6,Citation15,Citation16 To test the potential involvement of ERCC1 in these repair pathways in our cell models, we measured the ability of ERCC1 isoforms to bind to FANCG and SLX4 in PLA. ERCC1 isoform 202 gives the strongest signal in PLA when assessing interaction with FANCG or SLX4 (). The unique ability of isoform 202 to interact with these partners is in line with its ability to rescue the multinucleated phenotype described previously. These data therefore reveal a possible mechanism involving isoform 202 in the resolution of DNA structures generated by replication stress, ICLs, and DSB, which can cause anaphase bridges if incompletely processed. Consequently, the loss of the ERCC1 isoform 202 can result in abnormal mitosis and lead to aneuploidy and multinucleation.
The ERCC1–202 isoform mediates cell survival to mitomycin C
ICL-R in proliferating cells involves the FA pathway and homology-directed repair. To functionally test the relevance of the unique ability of the ERCC1–202 isoform to interact with FANCG and SLX4, we investigated the sensitivity of the ERCC1-deficient and single isoform-complemented cells to the ICL-generating drug Mitomycin C. ERCC1-deficient cells were more sensitive to the drug than the WT cells (, IC50 0.51 µM vs. 1.42 µM, respectively). Moreover, isoform 202 was the only isoform able to rescue the ERCC1-deficiency-induced sensitivity to mitomycin C (). These data confirm isoform 202 is the only ERCC1 product active in pathways processing ICLs and, as such, involved in cell resistance to chemotherapy.
Lack of dominant-negative isoform
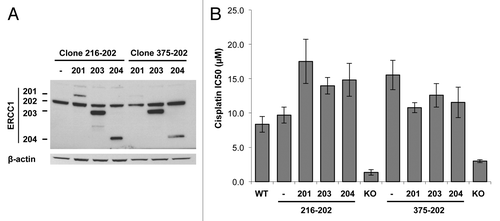
These results, together with previous data,Citation20 support that isoform 202 is the only product of ERCC1 active not only in NER but also in FA and homology-directed pathways. Another function has nevertheless been suggested for the ERCC1 isoform 203 as a dominant-negative modulator of ERCC1 activity.Citation29-Citation31 We therefore searched for dominant-negative effects of the ERCC1 isoforms toward the active isoform 202. For that purpose, two A549 ERCC1-deficient clones were successively transduced with a lentiviral vector expressing isoform 202, and then additionally with a lentivirus driving the expression of either isoform 201, 203, or 204. This strategy generated cell clones expressing the functionally active isoform 202 alongside with one of the other isoforms (). We then assessed the effect of each isoform combination on the 202-driven resistance to cisplatin cytotoxicity (). In this experimental setting, we did not observe any decrease of isoform 202 function when reintroducing either of the other isoforms. This suggests that ERCC1 isoforms 201, 203, and 204 do not have dominant-negative activity on ERCC1 202 isoform toward the repair of cisplatin adducts.
Figure 6. Absence of dominant-negative activity of ERCC1–201, 203 and 204 isoforms toward ERCC1–202. ERCC1-deficient A549 clones were complemented with the ERCC1–202 isoform (clones 216–202 and 375–202) and then transduced with a lentiviral vector driving the expression of each remaining isoforms. (A) Immunoblot analysis of ERCC1 expression. β-actin detection was used as a loading control. (B) Sensitivity of A549 cells expressing ERCC1–202 along with one of the remaining ERCC1 isoform to a 48h cisplatin treatment was determined by WST-1 assay. The average IC50 values from 2 independent experiments were plotted. Error bars indicate SEM.
Discussion
In this study, we investigated the role of each known ERCC1 isoform in the various DNA repair pathways involving the ERCC1/XPF endonuclease. We confirmed that isoform 202 is the only ERCC1 product active in NER and able to stabilize XPF. We also revealed the specific role of isoform 202 in ICL-R by both biochemical and functional assays.
Our results first demonstrate at a functional level what had been previously suggested by structural analysis of the ERCC1 isoforms. ERCC1 isoform 202 is the only isoform that encompasses the full XPA, XPF, and RPA-binding domains and was found to account for all ERCC1 DNA-damage response (DDR)-related functions in our study. The absence of activity of isoform 201 can be explained by its modified C-terminal end, a part of the protein that has proven to be required for ERCC1 functionality in NER.Citation32 Isoform 203 lacks exon 8, which encodes part of the HhH2 (Helix-hairpin-Helix) domain of ERCC1 that is involved in its interaction with XPF. Our results also showed the inability of isoform 204 to interact with XPF. This isoform lacks exon 3, which encodes the N-terminal part of the ERCC1 central domain that mediates interaction with XPA. We indeed found a decreased ability of this isoform to interact with XPA in PLA (). Even though the partial truncation of this domain may explain the absence of activity of isoform 204 in NER, there is no obvious structural basis for the inability of this isoform to interact with XPF, and a more global structural perturbation of the protein could possibly impact on the HhH2 domain function. Overall, the inability of the 201, 203, and 204 isoforms to interact with XPF is in line with their lack of activity in NER, ICL-R, and HDR, as these pathways require the endonuclease activity of the complex.
A dominant-negative role for the 203 isoform has been previously proposed.Citation29-Citation31 We could not find any evidence supporting this hypothesis, at least with regards to the DDR-related function of ERCC1 after inducing DNA damage with platinum agents. Cisplatin generates several types of DNA adduct that are processed by distinct DNA repair pathways and differently impact cell survival. Current assays do neither allow to discriminate between these distinct pathways, nor to evaluate their individual contribution on cell survival. It is conceivable that a dominant-negative activity of isoform 203—or of any isoform—would be pathway-dependent, and that the collective evaluation of several DNA repair pathways would preclude from detecting such activity. Moreover, this potential dominant-negative activity may vary according to the cell line model and the DDR profile of this latter. Whether any isoform displays a dominant-negative activity should therefore ideally be addressed by considering individually each DNA repair pathway in which ERCC1 is involved.
Interestingly, ERCC1 was not only detected in the nucleus, but also in the cytoplasm (notably for isoform 203), suggesting alternative functions or partners of ERCC1 that would not directly be linked to DDR. Also, we observed that isoforms 201, 203, and 204 isoforms could form nuclear foci but were unable to interact with XPF ( and ). This suggests that these isoforms are also involved in DDR and can interact with DNA repair factors, which still remain to be determined. Even though such an XPF-independent role of ERCC1 has not been reported yet, further investigation is warranted to decipher the role of these ERCC1 isoforms.
ERCC1-deficient cells displayed high rate of multinucleation. We propose that the incomplete processing of DNA lesions occurring in S and G2 phase of the cell cycle, due to abnormal activity of XPF/ERCC1, leads to the formation of anaphase DNA bridges that subsequently result in cytokinesis failure and aneuploidy. Several lines of evidence support this model. First, ERCC1 has been shown to interact with the FA pathway proteins FANCG and SLX4 (FANCP).Citation14-Citation16 These interactions indeed have functional relevance, since the nuclease activity of XPF/ERCC1 is required for the processing of ICL by the FA pathway.Citation3 Second, defective FA pathway can result in the formation of anaphase bridges.Citation27,Citation28 Third, the presence of anaphase bridges during mitosis in FA-deficient pathway cells can lead to cytokinesis failure and aneuploidy.Citation26 Finally, recent work has demonstrated a functional interplay between ERCC1/XPF and FANCD2 on mitotic chromosomes, both proteins interacting together in order to protect common fragile sites and avoid anaphase bridges and subsequent chromosomal instability.Citation33 Of note, as interwined chromatids may lead to gross chromosome loss or rearrangement, we investigated whether ERCC1-deficient A549 cells displayed genomic instability by performing CGH and cytogenetic analysis of the ERCC1-WT cell line and the ERCC1-deficient clones. We did not observe chromosomal rearrangements in our cell line models, supporting that the main outcome of anaphase bridges may be cytokinesis failure (data not shown).
Taken together, our data suggest that the ERCC1-isoform 202 fulfills all the clinically relevant functions of ERCC1, most likely because of its unique ability to interact with XPF. As it is not currently possible to distinguish between the ERCC1 isoforms in clinical samples, the detection of the XPF/ERCC1 heterodimer may represent a reliable surrogate biomarker for assessing ERCC1 functionality. The feasibility of such a strategy in cell lines models and patient samples is currently under investigation.
Materials and Methods
Cell lines and proliferation assays
Two clones of A549 ERCC1-deficient cells 216 and 375 were tested to rescue ERCC1 isoform expression and assess ERCC1 functions.Citation20 Cells were grown in DMEM media (Gibco-Invitrogen) supplemented with 10% fetal calf serum (FCS). The cell proliferation was determined in a short-term assay based on the reduction of WST-1 (water-soluble tetrazolium salt) (Roche Molecular) after 48 h of treatment with various concentrations of cisplatin (from 0.2 to 40 µM) and mitomycin-C (from 0.75 to 100 μM) and the IC50 was determined.
Cell cycle and DNA content
To study effect of cisplatin on cell cycle arrest, cells were treated with 30 nM or 300 nM of cisplatin for 48 h. For high DNA content analysis, cells were blocked in G2/M cell cycle phase with Karyomaxcolcemid solution (Gibco-Invitrogen) at 0.1 µg/ml for 6 h. DNA content was determined in ethanol-fixed cells, stained with propidium iodide, and analyzed using a Becton Dickinson FACScalibur flow cytometer and the CellQuest Pro software.
Western blot analysis
Proteins were extracted by lysis in RIPA buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholic acid, 0.5% NP-40, 0.1% SDS) supplemented with a protease inhibitor cocktail (Complete; Roche Molecular). For nucleus and cytoplasm protein, a first extraction and separation was done with a buffer containing 10 mM HEPES, 10 mMKCl, 1 mM DTT, 1 mM PMSF and protease inhibitor cocktail supplemented with 0.3% NP40. The nucleus fraction was next resuspended in a buffer containing 20 mM HEPES and 400 mM NaCl. Protein were then separated by SDS-PAGE and transferred to nitrocellulose membranes by the iBlot® 7-Minute Blotting System (Invitrogen). Blots were incubated with primary and secondary peroxidase-conjugated antibodies, and chemiluminescent detection was done using the Dura HRP Substrate (Thermo Scientific).
The antibodies used were ERCC1–3H11 (sc53281; Santa Cruz,), ERCC1–8F1 (MS-671P1; MM France), XPF-3F2/3 (Ab85140; Abcam), FANCG (Ab54645; Abcam), SLX4 (H00084464; Abnova), XPA (MMA1–21460; Pierce), MSH2 (orb16010; BIORBYT), β-actin (A5441, Sigma-Aldrich), and β-tubulin (T8328, Sigma-Aldrich) antibodies.
Histology staining
For cells morphology study, cells were fixed and stained with Diff Quik Kit (130832; DadeBehring/ Siemens) according to the manufacturer’s instructions.
For time-lapse experiment, cells were first stained with cell tracker green at 2.5 µM for 30 min (C2925; Invitrogen) and then stained with Hoechst 1/8000 (62249SPCL; thermo scientific). Images were acquired using a confocal laser scanning microscope (Fluoview SV10i - Olympus)
α and γ tubulin immunofluorescence staining
Microtubules were first stabilized in PHEM buffer, and then cells were fixed and permeabilized in cold methanol for 5 min. After washing with PBS 0.1% Tween, and with IFF buffer (PBS, BSA 2%, FCS 5%), cells were incubated with primary antibody (1:200 for β-tubulin antibody (T8328; Sigma) and 1:1000 for γ-tubulin [Sigma, T5192]) in IFF (PBS, 1% BSA, 2% FCS) for 45 min at room temperature. Cells were washed with PBS 0.1% Tween and incubated with secondary fluorescent antibody Alexafluor (Invitrogen) in IFF for 30 min at room temperature. After washing with PBS 0.1% Tween, slides were mounted with AntifadeProLong with DAPI (Invitrogen).
ERCC1-XPF immunofluorescence staining
Cells were fixed and permeabilized in formol and SDS 0.1% and then washed with PBS. After blocking with BSA 5%, cells were incubated with primary antibodies (1:200) ERCC1-FL297 (sc-10785; Santa Cruz), XPF-3F2/3 (ab85140; Abcam) in blocking solution for 1 h at 37 °C. Cells were washed with PBS and incubated with secondary fluorescent antibody Alexafluor (Invitrogen) in blocking solution for 1 h at 37 °C. After washing with PBS, slides were mounted with AntifadeProLong with DAPI (Invitrogen).
Proximity ligation assay (PLA)
Protein interactions were studied using the Duolink® II proximity ligation assays (PLA) kit (Olink Bioscience). Coverslips were processed according to the manufacturer’s instructions. In brief, the cells were fixed with methanol, stained with the primary antibodies, and then incubated with the secondary oligonucleotide-linked antibodies. The oligonucleotides were hybridized, ligated, amplified, and detected using a fluorescent probe.
For all IF staining, images were acquired with an Inverted Ti-E fluorescence microscope (Nikon) and were processed with ImageJ software. Each fluorescent signal dot was considered a single ERCC1 complex formation. The number of dots was normalized by the total nuclei area.
Acknowledgments
This work was supported by grants from Institut National du Cancer (PNES Poumon), Paris Sud University (Attractivité 2012), and Fondation de France. L Friboulet’s post-doc was funded by a Roche translational research fellowship.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Usanova S, Piée-Staffa A, Sied U, Thomale J, Schneider A, Kaina B, Köberle B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol Cancer 2010; 9:248; http://dx.doi.org/10.1186/1476-4598-9-248; PMID: 20846399
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol 2004; 24:5776 - 87; http://dx.doi.org/10.1128/MCB.24.13.5776-5787.2004; PMID: 15199134
- Bhagwat N, Olsen AL, Wang AT, Hanada K, Stuckert P, Kanaar R, D’Andrea A, Niedernhofer LJ, McHugh PJ. XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol Cell Biol 2009; 29:6427 - 37; http://dx.doi.org/10.1128/MCB.00086-09; PMID: 19805513
- Kikuchi K, Narita T, Pham VT, Iijima J, Hirota K, Keka IS, Mohiuddin, Okawa K, Hori T, Fukagawa T, et al. Structure-specific endonucleases Xpf and Mus81 play overlapping but essential roles in DNA repair by homologous recombination. Cancer Res 2013; 73:4362 - 71; http://dx.doi.org/10.1158/0008-5472.CAN-12-3154; PMID: 23576554
- Motycka TA, Bessho T, Post SM, Sung P, Tomkinson AE. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J BiolChem 2004; 279:13634 - 9; http://dx.doi.org/10.1074/jbc.M313779200; PMID: 14734547
- Al-Minawi AZ, Lee YF, Håkansson D, Johansson F, Lundin C, Saleh-Gohari N, Schultz N, Jenssen D, Bryant HE, Meuth M, et al. The ERCC1/XPF endonuclease is required for completion of homologous recombination at DNA replication forks stalled by inter-strand cross-links. Nucleic Acids Res 2009; 37:6400 - 13; http://dx.doi.org/10.1093/nar/gkp705; PMID: 19713438
- Al-Minawi AZ, Saleh-Gohari N, Helleday T. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res 2008; 36:1 - 9; http://dx.doi.org/10.1093/nar/gkm888; PMID: 17962301
- McNeil EM, Melton DW. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res 2012; 40:9990 - 10004; http://dx.doi.org/10.1093/nar/gks818; PMID: 22941649
- Li L, Elledge SJ, Peterson CA, Bales ES, Legerski RJ. Specific association between the human DNA repair proteins XPA and ERCC1. ProcNatlAcadSci U S A 1994; 91:5012 - 6; http://dx.doi.org/10.1073/pnas.91.11.5012; PMID: 8197174
- de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev 1998; 12:2598 - 609; http://dx.doi.org/10.1101/gad.12.16.2598; PMID: 9716411
- Bessho T, Sancar A, Thompson LH, Thelen MP. Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J BiolChem 1997; 272:3833 - 7; http://dx.doi.org/10.1074/jbc.272.6.3833; PMID: 9013642
- Fisher LA, Bessho M, Wakasugi M, Matsunaga T, Bessho T. Role of interaction of XPF with RPA in nucleotide excision repair. J MolBiol 2011; 413:337 - 46; http://dx.doi.org/10.1016/j.jmb.2011.08.034; PMID: 21875596
- Matsunaga T, Park CH, Bessho T, Mu D, Sancar A. Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nuclease. J BiolChem 1996; 271:11047 - 50; http://dx.doi.org/10.1074/jbc.271.19.11047; PMID: 8626644
- Wang C, Lambert MW. The Fanconi anemia protein, FANCG, binds to the ERCC1-XPF endonuclease via its tetratricopeptide repeats and the central domain of ERCC1. Biochemistry 2010; 49:5560 - 9; http://dx.doi.org/10.1021/bi100584c; PMID: 20518486
- Svendsen JM, Smogorzewska A, Sowa ME, O’Connell BC, Gygi SP, Elledge SJ, Harper JW. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009; 138:63 - 77; http://dx.doi.org/10.1016/j.cell.2009.06.030; PMID: 19596235
- Muñoz IM, Hain K, Déclais AC, Gardiner M, Toh GW, Sanchez-Pulido L, Heuckmann JM, Toth R, Macartney T, Eppink B, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell 2009; 35:116 - 27; http://dx.doi.org/10.1016/j.molcel.2009.06.020; PMID: 19595721
- Rageul J, Frëmin C, Ezan F, Baffet G, Langouët S. The knock-down of ERCC1 but not of XPF causes multinucleation. DNA Repair (Amst) 2011; 10:978 - 90; http://dx.doi.org/10.1016/j.dnarep.2011.07.005; PMID: 21839691
- Tan LJ, Saijo M, Kuraoka I, Narita T, Takahata C, Iwai S, Tanaka K. Xerodermapigmentosum group F protein binds to Eg5 and is required for proper mitosis: implications for XP-F and XFE. Genes Cells 2012; 17:173 - 85; http://dx.doi.org/10.1111/j.1365-2443.2012.01582.x; PMID: 22353549
- Olaussen KA, Dunant A, Fouret P, Brambilla E, André F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, et al, IALT Bio Investigators. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med 2006; 355:983 - 91; http://dx.doi.org/10.1056/NEJMoa060570; PMID: 16957145
- Friboulet L, Olaussen KA, Pignon JP, Shepherd FA, Tsao MS, Graziano S, Kratzke R, Douillard JY, Seymour L, Pirker R, et al. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N Engl J Med 2013; 368:1101 - 10; http://dx.doi.org/10.1056/NEJMoa1214271; PMID: 23514287
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protić M, Hübscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 1995; 80:859 - 68; http://dx.doi.org/10.1016/0092-8674(95)90289-9; PMID: 7697716
- Arora S, Kothandapani A, Tillison K, Kalman-Maltese V, Patrick SM. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair (Amst) 2010; 9:745 - 53; http://dx.doi.org/10.1016/j.dnarep.2010.03.010; PMID: 20418188
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. CurrBiol 1997; 7:427 - 39; http://dx.doi.org/10.1016/S0960-9822(06)00190-4; PMID: 9197240
- Melton DW, Ketchen AM, Núñez F, Bonatti-Abbondandolo S, Abbondandolo A, Squires S, Johnson RT. Cells from ERCC1-deficient mice show increased genome instability and a reduced frequency of S-phase-dependent illegitimate chromosome exchange but a normal frequency of homologous recombination. J Cell Sci 1998; 111:395 - 404; PMID: 9427687
- Chipchase MD, Melton DW. The formation of UV-induced chromosome aberrations involves ERCC1 and XPF but not other nucleotide excision repair genes. DNA Repair (Amst) 2002; 1:335 - 40; http://dx.doi.org/10.1016/S1568-7864(02)00010-1; PMID: 12509251
- Vinciguerra P, Godinho SA, Parmar K, Pellman D, D’Andrea AD. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J Clin Invest 2010; 120:3834 - 42; http://dx.doi.org/10.1172/JCI43391; PMID: 20921626
- Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 2009; 11:761 - 8; http://dx.doi.org/10.1038/ncb1883; PMID: 19465921
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 2009; 11:753 - 60; http://dx.doi.org/10.1038/ncb1882; PMID: 19465922
- Dabholkar M, Vionnet J, Parker R, Bostickbruton F, Dobbins A, Reed E. Expression of an alternatively spliced ercc1 messenger-RNA species, is related to reduced DNA-repair efficiency in human T-lymphocytes. Oncol Rep 1995; 2:209 - 14; PMID: 21597714
- Yu JJ, Mu C, Dabholkar M, Guo Y, Bostick-Bruton F, Reed E. Alternative splicing of ERCC1 and cisplatin-DNA adduct repair in human tumor cell lines. Int J Mol Med 1998; 1:617 - 20; PMID: 9852275
- Sun Y, Li T, Ma K, Tian Z, Zhu Y, Chen F, Hu G. The impacts of ERCC1 gene exon VIII alternative splicing on cisplatin-resistance in ovarian cancer cells. Cancer Invest 2009; 27:891 - 7; http://dx.doi.org/10.3109/07357900902744536; PMID: 19832035
- Sijbers AM, van der Spek PJ, Odijk H, van den Berg J, van Duin M, Westerveld A, Jaspers NG, Bootsma D, Hoeijmakers JH. Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res 1996; 24:3370 - 80; http://dx.doi.org/10.1093/nar/24.17.3370; PMID: 8811092
- Naim V, Wilhelm T, Debatisse M, Rosselli F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 2013; 15:1008 - 15; http://dx.doi.org/10.1038/ncb2793; PMID: 23811686