
Abstract
Oncogene-induced senescence is a permanent cell cycle arrest characterized by extensive chromatin reorganization. Here, we investigated the specific targeting and dynamics of histone H3 variants in human primary senescent cells. We show that newly synthesized epitope-tagged H3.3 is incorporated in senescent cells but does not accumulate in senescence-associated heterochromatin foci (SAHF). Instead, we observe that new H3.3 colocalizes with its specific histone chaperones within the promyelocytic leukemia nuclear bodies (PML-NBs) and is targeted to PML-NBs in a DAXX-dependent manner both in proliferating and senescent cells. We further show that overexpression of DAXX enhances targeting of H3.3 in large PML-NBs devoid of transcriptional activity and promotes the accumulation of HP1, independently of H3K9me3. Loss of H3.3 from pericentromeric heterochromatin upon DAXX or PML depletion suggests that the targeting of H3.3 to PML-NBs is implicated in pericentromeric heterochromatin organization. Together, our results underline the importance of the replication-independent chromatin assembly pathway for histone replacement in non-dividing senescent cells and establish PML-NBs as important regulatory sites for the incorporation of new H3.3 into chromatin.
Keywords: :
Introduction
Most mammalian cells only divide a limited number of times before they undergo terminal differentiation or enter the state of senescence. Cellular senescence may be triggered by various forms of stress stimuli. First described as the result of replicative exhaustion of cultured normal diploid cells,Citation1 senescence can also be induced by oxidative stress, activated oncogenes such as H-RasV12, or inadequate growth conditions.Citation2-Citation5 Oncogene-induced senescence (OIS) results from a DNA damage response (DDR) activated by aberrant DNA replicationCitation6-Citation8 and may pose as an important anti-tumor barrier. Identification of senescent cells in benign or premalignant, but not malignant tissues or using various human and mouse model systems seems to support this hypothesis.Citation9-Citation13 Like terminal differentiation, senescence is characterized by irreversible cell cycle arrest and rigorous reorganization of cellular morphology, including the structure of the chromatin.
Chromatin is comprised of nucleosomes that each consists of 147 base pairs of DNA wrapped around a core histone octamer. The histone octamer is composed of a central (H3-H4)2 tetramer flanked by 2 H2A–H2B histone dimers.Citation14 Three principle mechanisms bring about chromatin alterations in eukaryotic cells: (1) post-translational modification of histone tails, (2) the action of chromatin remodeling enzymes, and (3) the replacement of canonical histone proteins by histone variants.Citation14 Incorporation of histone variants into chromatin is orchestrated by a family of proteins called histone chaperonesCitation15 and may provide different biophysical properties to the chromatin fiber or different post-translational modification sites, thus influencing nucleosome stability and function.Citation14,Citation16
Histone H3.3 is a variant of histone H3 that differs by only 5 amino acids from the canonical replicative histone variant H3.1 and has emerged as a regulator of chromatin states.Citation17 H3.3 is constitutively expressed throughout the cell cycle and in quiescenceCitation18 and is incorporated into chromatin in a DNA synthesis-independent manner.Citation19,Citation20 It is enriched within actively transcribed genes,Citation21-Citation25 but also accumulates at pericentromeric and telomeric heterochromatin regions.Citation26-Citation28 While the histone chaperone HIRA, along with associated factors, ASF1a, Ubinuclein1, and Cabin1, is responsible for H3.3 deposition into active chromatin,Citation19,Citation20,Citation27,Citation29-Citation32 the H3.3-specific chaperone DAXX in cooperation with the chromatin remodeler ATRX is essential for H3.3 deposition at heterochromatic loci.Citation26,Citation27,Citation33 The ATRX/DAXX/H3.3 pathway has been implicated in the suppression of pancreatic neuroendocrine tumors (panNET) and pediatric glioblastomas,Citation34-Citation39 thus establishing its role in carcinogenesis.
While establishment and maintenance of chromatin structure is central for genome function,Citation40 how such a mechanism is achieved in senescent cells has remained unclear. Chromatin structure is extensively remodeled upon senescence entry, as exemplified by the formation of senescence-associated heterochromatin foci (SAHF), visible as microscopically discernible, punctate DNA foci in DAPI-stained senescent cells.Citation41 These structures are thought to contribute to the senescence-associated cell cycle arrest in part by silencing proliferation-promoting genes through heterochromatinization.Citation41 Moreover, oncogene-induced SAHF formation may protect premalignant cells to undergo apoptosis by limiting extensive DNA damage to sub-lethal levels.Citation42
Little is known about the underlying mechanisms of the extensive chromatin reorganization observed in senescent cells. SAHF are enriched in markers of heterochromatin, including tri-methylated histone H3 at lysine 9 (H3K9me3), all HP1 isoforms, as well as HMGA proteins.Citation41,Citation43 In addition, SAHF are also enriched in the histone H2A variant macroH2A,Citation44 a variant associated with gene silencing, as, for example, during X inactivation.Citation45 Formation of MacroH2A- and HP1-containing SAHF is dependent on the 2 histone chaperones HIRA and ASF1a,Citation44 suggesting that H3.3 may become enriched in SAHF during OIS.Citation44,Citation46 Interestingly, SAHF formation also depends on the prior localization of HIRA into promyelocytic leukemia (PML) nuclear bodies (PML-NBs),Citation44,Citation47 discrete foci, 0.2–1.0 μm wide, that are present in most mammalian cell nuclei and stain positive for the tumor suppressor PML.Citation48,Citation49 PML-NBs have previously been implicated in the onset of OIS: they increase in number and size upon overexpression of H-RasV12, and overexpression of PML triggers p53-dependent senescence.Citation50,Citation51 Thus, PML-NBs may represent important regulatory structures not only for the induction of OIS in general, but also for the establishment and maintenance of the specialized chromatin structure characteristic of the senescent state.
In this study, we set out to investigate the dynamics of H3.3 incorporation in senescent cells, and, in particular, its possible connection with PML-NBs. To this end we employed the novel SNAP-tagging approach, which has been successfully used to investigate the deposition of newly synthesized H3 variants in human cells.Citation20,Citation52 Surprisingly, we show here that H3.3 is not enriched in SAHF. Instead, we find that H3.3 is incorporated in active chromatin regions during senescence. In addition, we also observe the DAXX-dependent recruitment of H3.3 into PML-NBs both in proliferating and senescent cells, thus establishing PML-NBs as important assembly points for newly synthesized H3.3 histones. We further show a decrease in the localization of H3.3 to satellite DNA regions upon depletion of DAXX or PML in human primary cells, thus linking the targeting of H3.3 to PML-NBs with the maintenance of pericentromeric heterochromatin.
Results
An in vivo visualization assay for newly synthesized H3.1 and H3.3 deposition in primary cells
In order to address the dynamics of the histone variants H3.1 and H3.3 in proliferating and senescent cells, we established human MRC5 primary diploid fibroblasts stably expressing H3.1 or H3.3 tagged with SNAP and 3 HA epitopes (MRC5 e-H3.1 and e-H3.3, respectively). SNAP is a modified variant of a suicide DNA repair enzyme that catalyzes its own irreversible covalent binding to the cell-permeable molecule benzylguanine (BG). For in vivo labeling assays, BG is replaced by fluorescent or non-fluorescent derivatives.Citation53,Citation54 This technique is tailor-made to visualize proteins synthesized at different time points, thus permitting to distinguish between old histone proteins and newly synthesized onesCitation20,Citation52,Citation54 (Fig. S1A). In particular, this system proved essential to address the incorporation and localization of new histones in senescent cells, because in regular epitope-tagged H3 expressing cell lines, it would be impossible to distinguish between incorporation of new histones vs. chromatin-wide incorporation of H3 after senescence induction.
We first verified by immunofluorescent staining that the tagged histones colocalize with mitotic chromosomes, indicating that the presence of the epitope tag does not interfere with the deposition of H3.1 nor H3.3 into chromatin in vivo (Fig. S1B). We then validated in both cell lines the efficiency of our in vivo deposition assay based on quench–chase–pulse (QCP) labeling. Microscopic analysis shows that the pulse with TMR-Star labeled both pre-existing H3.1 and H3.3 efficiently. In contrast, although pre-existing histones could still be labeled with HA antibody, a quench–pulse assay only gave background staining confirming the efficiency of quenching. A chase period during which new histone biosynthesis takes place then allowed the selective labeling of the new histones with TMR-Star (; Fig. S1A–C). Furthermore, by combining quench–chase–pulse labeling assay with BrdU incorporation to detect replicating cells, we confirmed that H3.1 deposition is limited to S-phase, while H3.3 deposition occurs throughout the cell cycle (Fig. S1B–D).Citation20 We conclude that human primary MRC5 cell lines expressing SNAP-HAx3-tagged histones are a powerful tool to analyze the mechanisms of de novo deposition of H3.1 and H3.3 at the single-cell level.
Figure 1. Dynamics of H3.1 and H3.3 deposition in proliferating and senescent human cells. (A) Fluorescent microscopy visualization of H3.1- and H3.3-SNAP-HAx3 after in vivo labeling assays of MRC5 human primary cells with red fluorescent TMR-Star in pulse, quench–pulse, and quench–chase–pulse experiments. The pulse labels pre-existing H3-SNAP, the quench-pulse quenches pre-existing H3-SNAP with nonfluorescent block preventing their subsequent labeling with TMR-Star (background), and the quench–chase–pulse labels new H3-SNAP synthesized during the 3h30 chase. In all cases, HA stains total histone H3. DAPI stains nuclei. Scale bar is 10 μm. (See also Fig.S1). (B) Western blot analysis of whole cell extracts from MRC5 stably expressing H3.1- or H3.3 SNAP-HAx3 (e-H3.1 and e-H3.3, respectively) and transduced with an empty retroviral vector (empty) or with a vector expressing H-RasV12 (Ras) for 10 d. 25 μg of protein extracts were loaded. HA and Ras stainings verified expression of the transduced proteins. Mcm7 was used as a marker for cell proliferation and p16 as a marker for proliferation arrest. Smc-1 served as a loading control. M, molecular weight marker. (See also Fig.S2A–C). (C) Fluorescent microscopy visualization of new H3.1 and H3.3 (TMR, red) after in vivo labeling of MRC5 e-H3.1 and e-H3.3 treated as in (B) in a quench–chase–pulse experiment. HA (green) stains total H3 histones and DAPI stains nuclei. Scale bar is 10 μm. (See also Fig.S2D). (D) Histogram shows quantitative analysis of the proportion of TMR-positive cells in MRC5 e-H3.1 or e-H3.3 proliferating (empty) and senescent (Ras) cells. Numbers represent the mean of 3 independent experiments ± s.d.
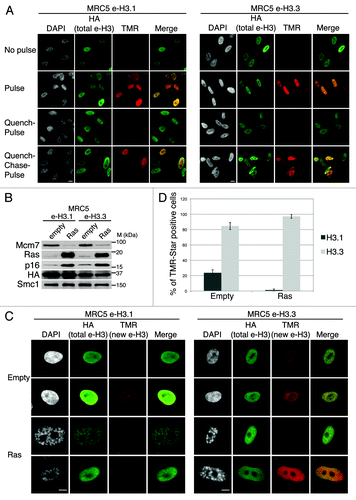
Newly synthesized H3.3 is actively deposited in senescent cells, while H3.1 is not
To analyze the deposition of H3.1 and H3.3 variants in senescent cells, we induced senescence in MRC5 human primary diploid cells by overexpression of oncogenic Ras using retroviral-mediated gene transfer. At 8 d post-infection, MRC5 cells had arrested at sub-confluent density, displayed reduced BrdU incorporation, and showed the characteristic formation of SAHF visible as DAPI-dense foci, as described previously (Fig. S2A and C).Citation4,Citation41 In addition, MRC5 cells also stained positive for the senescence-associated β-galactosidase,Citation55,Citation56 thus confirming the entry into senescence (Fig. S2B and C). We then overexpressed oncogenic Ras to induce senescence in MRC5 e-H3.1 or e-H3.3 cells () and performed quench–chase–pulse experiments to follow new histone deposition into chromatin. We verified that the newly synthesized histones are incorporated into chromatin by performing a detergent extraction to remove soluble histones prior to fixation of the cells (Fig. S2D). Newly deposited H3.1 was detected in about 24% of proliferating cells (), corresponding to S-phase cells (Fig. S1C and D), consistent with previous work.Citation19,Citation20 In contrast, new H3.1 was not incorporated in senescent cells, even though we could still detect the pool of pre-existing histones H3.1 by HA staining (). Interestingly, pre-existing e-H3.1 accumulated at SAHF regions, marked as H3K9me3/DAPI-dense foci, possibly as a result of the high condensation of these heterochromatin regions (). We next examined the deposition of new H3.3 in proliferating primary cells and confirmed its incorporation throughout the cell cycle in about 85% of cells. This number did not reach 100%, because a certain percentage of cells expressed very low levels of recombinant histones, which resulted in undetectable TMR-Star signal. Importantly, we could show for the first time de novo deposition of new H3.3 in senescent cells (). Quantification of the number of TMR-positive cells in Ras-arrested cells revealed that almost all senescent cells incorporated new H3.3 (). Of note, endogenous H3.3 is still efficiently expressed in senescent cells (at about 60% of its levels in proliferating cells), as seen by quantitative PCR, as compared with a reduction to about 35% for H3.1 (Fig. S3A and B), underscoring the importance to maintain a pool of H3.3 histones during senescence. Thus, our results suggest that replication-independent nucleosome assembly is the predominant mode of histone replacement in senescent cells.
Figure 2. New H3.3 deposition does not occur into SAHF, in contrast to parental H3.1, but correlates with transcriptional activity in human senescent cells. (A) Fluorescent microscopy visualization of pre-existing H3.1 (HA, green) in MRC5 e-H3.1 cells proliferating (empty) or induced into senescence with H-RasV12 overexpression (Ras) for 8 d. H3K9me3 (red) and DAPI were used as markers for SAHF formation. Scale bar is 10 μm. (B) Graphics show fluorescent intensity profiles quantified by ImageJ along lines drawn through nuclei as shown in (A). (C) Fluorescent microscopy visualization of nascent global RNA transcription in vivo in MRC5 cells treated as in (A). Nascent RNAs were labeled with 5-ethynyl uridine (EU) for 3 h and revealed by the Click-iT chemistry (red). H3K9me3 (green) and DAPI were used as markers for SAHF formation. Scale bar is 10 μm. (D) Graphics show fluorescent intensity profiles quantified using ImageJ along lines drawn through nuclei as shown in (C). (E) Fluorescent microscopy visualization of new H3.3 (TMR, red) after in vivo labeling of MRC5 e-H3.3 cells treated as in (A) in a quench–chase–pulse experiment. H3K9me3 (green) and DAPI were used as markers for SAHF formation. Scale bar is 10 μm. (See also Fig.S3CandD). (F) Graphics show fluorescent intensity profiles quantified by ImageJ along lines drawn through nuclei as shown in (E).
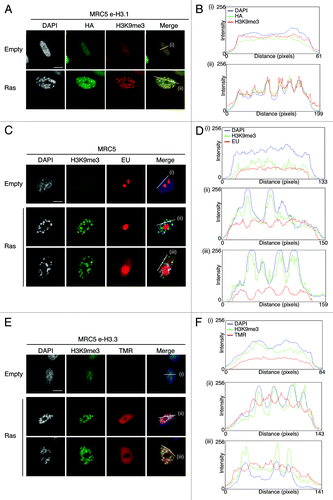
Newly synthesized H3.3 deposition in senescent cells correlates with active transcription
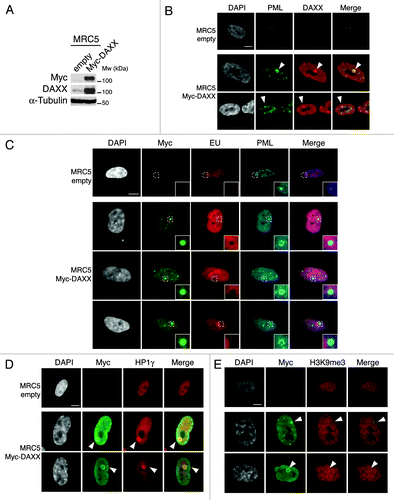
We then decided to examine more closely where exactly newly synthesized H3.3 is deposited in senescent cells. Given the difficulty to specifically detect endogenous H3 variants by microscopy (H3.1 and H3.3 differ in only 5 amino acids), it has not yet been possible to determine the histone H3 variant composition of SAHF. These foci were originally described as transcriptionally inactive heterochromatin on the basis of the presence of all HP1 isoforms as well as H3K9me3 by immunofluorescence analysis.Citation41,Citation44 We first set out to confirm the transcriptionally silent state of SAHF. By combining immunostaining of H3K9me3 to detect SAHF with 5-Ethynyl uridine (EU) to label global nascent RNA transcription in vivo, we observed that EU labeling was clearly excluded from SAHF, marked as H3K9me3/ DAPI-dense foci (). We quantified fluorescent intensity profiles along a line drawn through the centers of the nuclei of senescent cells and confirmed the inverse correlation between de novo RNA synthesis and SAHF presence in MRC5 senescent cells (). Thus, our results substantiate the transcriptionally silent state of the SAHF regions.
Importantly, SAHF formation is in part regulated by the H3.3 histone chaperones HIRA and ASF1a,Citation44,Citation47 suggesting involvement of the H3.3 histone variant in this process. We thus investigated if new H3.3 becomes incorporated in SAHF by using our powerful in vivo deposition assay. We combined a quench–chase–pulse labeling of new H3.3 by TMR-Star with H3K9me3 immunostaining as a marker of SAHF. We observed no enrichment of new H3.3 at H3K9me3/DAPI dense foci (), confirmed by the striking reverse correlation of fluorescent intensity profiles of new H3.3 (TMR) and SAHF (H3K9me3 and DAPI-dense peaks) (), reminiscent of the profiles observed with EU labeling. Confocal microscopy confirmed the absence of new H3.3 within SAHF in single plane images (Fig. S3C), thus strongly indicating that most incorporation of new H3.3 in senescent cells takes place at transcriptionally active regions of the nucleus. While epitope tagging can sometimes interfere with the function of the protein, we also extended these experiments to endogenous H3.3 by using a specific H3.3 antibodyCitation26 for immunofluorescence analysis in IMR90 ER:Ras primary fibroblasts driven into senescence with addition of 4-hydroxytamoxifen (4-OHT)Citation57 (Fig. S3D). While the endogenous H3.3 behaved similarly to SNAP-HA-tagged recombinant H3.3, our data do not exclude the possibility that H3.3 could also be deposited in specific heterochromatic regions outside of SAHF.
Newly synthesized H3.3 localizes in PML-NBs together with DAXX and ATRX in proliferating and senescent cells
We next inspected more closely H3.3 labeling patterns in senescent cells. Remarkably, we noticed a distinct intranuclear focal H3.3 staining in the vast majority of cells, but not in MRC5 e-H3.1 cells, suggesting a distinct chromatin assembly pathway for H3.1 and H3.3. Interestingly, the distinct nuclear H3.3 foci were also present in proliferating cells, although they were smaller in size and intensity as compared with senescent cells, reminiscent of the pattern observed for PML-NBs.Citation50,Citation51 We thus wondered if these H3.3 foci would co-localize with PML-NBs, together with its specific chaperones DAXX, ATRX, HIRA, and ASF1a. Combined quench–chase–pulse labeling of new H3.3 by TMR-Star with PML and DAXX/ATRX triple immunostaining clearly revealed that H3.3 co-localizes with PML-NBs in both proliferating and oncogene-induced senescent cells together with its associated histone chaperones DAXX and ATRX (). We confirmed these data by fluorescent intensity profiles drawn through nuclei (). We extended our results obtained on e-H3.3 to the endogenous histone H3.3 and showed its localization at PML-NBs both in proliferating and senescent human primary fibroblasts (Fig. S4A). We also observed increased size and intensities of DAXX, ATRX, and new H3.3 foci in senescent cells (; Fig. S4A), as demonstrated previously for PML-NBs.Citation50,Citation51 In addition, new H3.3 also co-localized with HIRA and ASF1a (Fig. S4B).
Figure 3. New H3.3 localizes in PML-NBs together with DAXX and ATRX in proliferating and senescent cells. (A) Fluorescent microscopy visualization of new H3.3 (TMR, red) after in vivo labeling of proliferating (empty) or senescent (Ras) MRC5 e-H3.3 cells in a quench–chase–pulse experiment. Co-staining with DAXX (green) and PML (cyan, pseudo-color) shows colocalization of new H3.3 with its chaperone DAXX in PML-NBs. Merge images represent PML in blue, DAXX in green, and new H3.3 (TMR) in red. Scale bar is 10 μm. (See also Fig.S4AandB). (B) Graphics show fluorescent intensity profiles quantified using ImageJ along lines drawn through nuclei as shown in (A). (C) Fluorescent microscopy visualization of new H3.3 (TMR, red) after in vivo labeling of MRC5 e-H3.3 cells treated as in (A) in a quench–chase–pulse experiment. Co-staining with ATRX (green) and PML (cyan, pseudo-color) shows colocalization of new H3.3 with its chaperone ATRX in PML-NBs. Merge images represent PML in blue, ATRX in green, and new H3.3 (TMR) in red. Scale bar is 10 μm. (D) Graphics show fluorescent intensity profiles quantified by ImageJ along lines drawn through nuclei as shown in (C). (E) Immunoprecipitation performed against HA tag on 150 μg of nuclear cell extracts from regular MRC5 cells (negative control) or MRC5 e-H3.1 or e-H3.3. Cells were either proliferating (empty) or induced into senescence by overexpression H-RasV12 (Ras) for 8 d. Input is 10% of the immunoprecipitated material. Membranes were probed for HA as a control for IP, for Mcm7 as a marker of cell proliferation and negative control for IP, and for DAXX and ATRX. M, molecular weight marker.
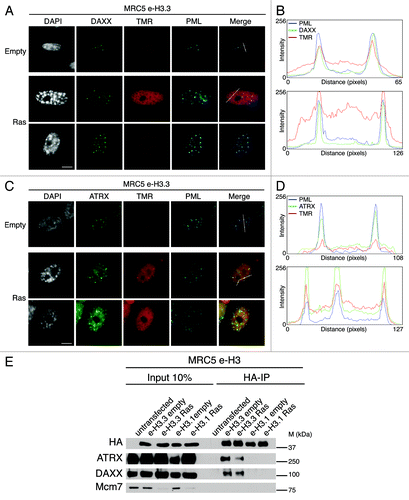
H3.3 interacts with DAXX and ATRX in proliferating cells.Citation26,Citation27,Citation33 Hence, the strong co-localization of e-H3.3 with DAXX and ATRX in PML-NBs in senescent cells prompted us to analyze the interaction of H3.3 with its chaperones in senescent cells. We first confirmed, by western blot analysis of fractionated cell extracts, that H3.3 as well as all H3.3 chaperones remain expressed in senescent MRC5 cells (Fig. S3B), in contrast to the proliferative marker MCM7 or to the H3.1 histone chaperone CAF-1.Citation58 We then purified histone complexes by HA immunoprecipitation in nuclear extracts from senescent MRC5 e-H3.1- and e-H3.3- expressing cells lines. Immunoblotting analysis of the purified complexes revealed the specific presence of DAXX and ATRX in H3.3 complexes, but not of H3.1 in senescent cells (), thus corroborating their function in H3.3 dynamics during senescence.
Interestingly, before the function of DAXX and ATRX in histone H3.3 chromatin assembly was identified, both factors were known to be components of PML-NBs in proliferating cellsCitation59,Citation60 raising the possibility that these chaperones could localize in PML-NBs during senescence to regulate replication-independent chromatin assembly. We thus sought to verify the localization of these H3.3 chaperones as well as HIRA and ASF1a in PML-NBs in absence of H3.3 overexpression. While only a subpopulation of cells showed HIRA and ASF1a at PML-NBs in proliferating cells, this number increased upon senescence, as described previously.Citation44,Citation61 In contrast, ATRX and DAXX co-localized each in PML-NBs in almost all proliferating and senescent cells (Fig. S4C). In addition, focal staining of all histone chaperones showed an increase in size and intensity in senescent cells, consistent with the pattern observed for PML-NBs.Citation50,Citation51 Of note, ATRX also localized to a fraction of SAHF in senescent cells, in addition to its localization to PML-NBs (Fig. S4C and S9H). Together, our results indicate that new H3.3 localizes to PML-NBs together with its associated chaperones, and that this occurs in an increased manner during senescence.
DAXX-dependent recruitment of H3.3 to PML-NBs
We next made use of our in vivo deposition assay to directly assess the role of histone chaperones in targeting of H3.3 to PML-NBs both in proliferating and senescent cells. Co-localization of DAXX and ATRX with new H3.3 at PML-NBs and their interaction with H3.3 in both proliferating and senescent cells () suggested that either chaperone could mediate H3.3 targeting to PML-NBs. To test this, we depleted DAXX and ATRX by RNA interference in MRC5 cells and first checked the efficiency of the knockdown by western blot analysis and ensured that depletion of each chaperone does not affect levels of the other (Fig. S5A). Strikingly, specific depletion of DAXX with 2 different siRNAs, but not of HIRA nor H3.3, led to a dramatic loss of ATRX from the PML-NBs (Fig. S5B)Citation60 and was accompanied by a striking increase of ATRX localization at SAHF in Ras-induced cells (Fig. S6A and B). In contrast, downregulation of ATRX, HIRA, or H3.3 did not affect DAXX localization in PML-NBs (Fig. S5B). Thus, ATRX localization in PML-NBs is DAXX-dependent.
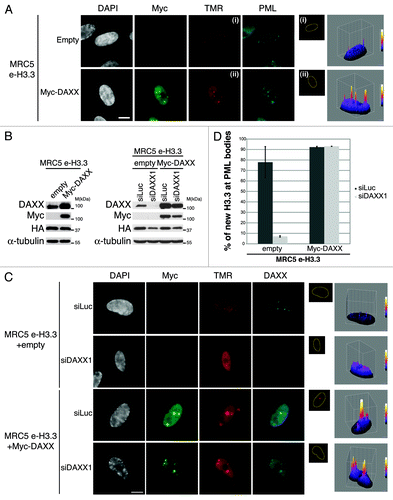
We then assessed the localization of newly synthesized H3.3 in PML-NBs in absence of DAXX or ATRX. To this end we first transduced MRC5 cells expressing e-H3.3 with empty vector or oncogenic Ras-expressing viruses followed by DAXX or ATRX depletion (). Efficient downregulation of the H3.3 chaperones and expression of oncogenic Ras were verified by western blot analysis (). We observed a dramatic loss of new H3.3 histones at PML-NBs upon DAXX depletion with 2 different sets of siRNAs, both in proliferating and in senescent cells (), confirmed by quantification (). As a control, PML depletion also led to a loss of H3.3 at PML-NBs (Fig. S7A–C). In contrast, neither knockdown of ATRX () nor HIRA (Fig. S7D–F) affected H3.3 localization at PML-NBs. Thus, our results reveal that DAXX promotes H3.3 recruitment to PML-NBs in proliferating and senescent cells.
Figure 4. New H3.3 is lost from PML-NBs in DAXX-depleted cells. (A) Scheme for the assay of in vivo labeling of new H3.3 in MRC5 e-H3.3 induced into senescence and depleted of specific histone H3.3 chaperones. (B) Western blot analysis of total cell extracts from MRC5 e-H3.3 treated as in (A). Membranes were probed for HA and Ras to verify expression of the transduced proteins, and for DAXX and ATRX. Smc1 served as a loading control. M, molecular weight marker. (C) Fluorescent microscopy visualization of new H3.3 (TMR, red) after in vivo labeling of MRC5 e-H3.3 cells treated as in (A) in a quench–chase–pulse experiment. Co-staining with PML (green) shows loss of new H3.3 at PML-NBs upon DAXX depletion. Insets represent enlarged images (3×) of selected area. Scale bar is 10 μm. (See also Fig.S7). (D) Histogram shows quantitative analysis of the proportion of cells showing new H3.3 localization at PML-NBs. Numbers represents the mean of 3 independent experiments ± s.d.
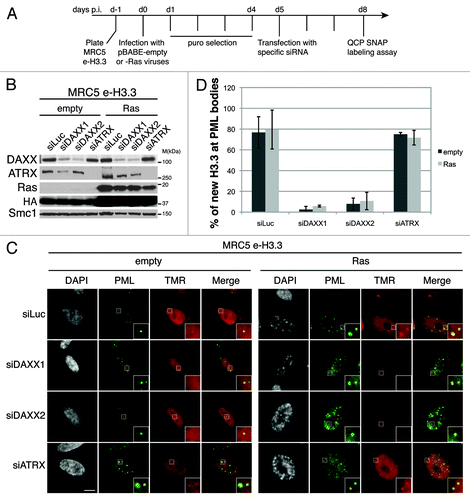
Given the essential role of PML in the induction of senescence through p53 activation and regulation of SAHF formation,Citation44,Citation50,Citation51 we wondered if DAXX, ATRX, or H3.3 depletion would impair Ras-induced senescence. We used 2 strategies to deplete DAXX and ATRX by shRNA. We either cotransduced MRC5 human primary fibroblasts with viruses encoding oncogenic Ras and the specific shRNA or we used IMR90 ER:Ras transduced with shRNAs 5 d before induction of senescence by addition of 4-OHT. In both cases, we could not detect any defects in SAHF formation or senescence entry (Figs. S8A–C and S9A-E). Of note, overexpression of DAXX did not affect oncogene-induced senescence either, because we observed normal reduction in Cyclin A levels, increase in p16 expression, and normal SAHF formation in MRC5 and IMR90 ER:Ras overexpressing DAXX and the oncogenic form of Ras (Figs. S8D and E and S9E). In addition, we also depleted DAXX, ATRX, and H3.3 by siRNA in IMR90 ER:Ras 1 d before the induction of senescence. After 6 d of 4-OHT and 2 rounds of siRNA transfection to ensure continuous knockdown of the proteins, we could not detect any defects in senescence entry in DAXX-, ATRX-, and H3.3-depleted cells (Figs. S9F–H and S4A). Thus, we decided to pursue our analysis in proliferating cells in order to investigate further the possible functions of H3.3 localization in PML-NBs.
DAXX overexpression increases localization of H3.3 at PML-NBs
We first asked if overexpression of DAXX protein would trigger the reverse phenotype as observed upon DAXX depletion. We therefore transduced MRC5 e-H3.3 cells with retroviruses encoding Myc-DAXX. Upon overexpression of DAXX, triple immunostaining analysis showed perfect colocalization of new H3.3 with Myc-DAXX in PML-NBs along with a marked increase of the H3.3 signal within PML-NBs (), sometimes leading to a massive accumulation of new H3.3 in a very large single PML-NB surrounded by a thin spherical enrichment of PML and DAXX proteins (Fig. S10A). We then assessed if overexpression of recombinant DAXX could rescue the loss of H3.3 at PML-NBs in proliferating cells that were depleted from endogenous DAXX. To this end, we generated a siRNA-resistant derivative of Myc-DAXX by inserting silent mutations in the siRNA-target region and verified that it is not targeted by the siDAXX1 (). Quench–chase–pulse labeling assays in MRC5 e-H3.3 cells depleted of endogenous DAXX showed that overexpression of Myc-DAXX rescues the localization of H3.3 in PML-NBs () and even leads to an increased H3.3 recruitment to PML-NBs, similar to the results obtained after overexpression of DAXX (). Together, our results underline the essential role of DAXX for the recruitment of H3.3 to PML-NBs and rule out an off-target effect of our DAXX siRNA.
Figure 5. Overexpression of DAXX increases new H3.3 localization at PML-NBs (A) (Left panel) Fluorescent microscopy visualization of new H3.3 (TMR, red) after in vivo labeling of MRC5 e-H3.3 cells in a quench–chase–pulse experiment. MRC5 e-H3.3 cells transduced with an empty vector (Empty) or a vector expressing Myc-DAXX (Myc-DAXX) were co-stained with Myc (green) and PML (cyan, pseudo-color). Scale bar is 10 μm. (Right panel) Graphics show distribution of new H3.3 (TMR) signal in MRC5 e-H3.3. TMR fluorescence intensity of each pixel delimited within the nuclei is plotted in a 3D-surface plot and shows increase in the targeting of new H3.3 at PML-NBs upon DAXX overexpression. (See also Fig.S10A). (B) Western blot analysis of total cell extracts from MRC5 e-H3.3 treated as in (A) or as in (C). Membranes were probed for HA and Myc to verify expression of the transduced proteins, and for DAXX. α-Tubulin is a loading control. M, molecular weight marker. (C) (Left panel) Fluorescent microscopy visualization of new H3.3 (TMR, red) after in vivo labeling of MRC5 e-H3.3 cells in a quench–chase–pulse experiment. Control MRC5 e-H3.3 cells and MRC5 e-H3.3 cells co-expressing a siRNA resistant form of Myc-DAXX were transfected for 2 d with the indicated siRNAs before labeling with TMR and co-staining with Myc (green) and DAXX (cyan, pseudo-color). Scale bar is 10 μm. (Right panel) Graphics showing distribution of new H3.3 (TMR) signal intensity as in (A), reveals rescue of new H3.3 localization to PML-NBs upon re-expression of DAXX. (D) Histogram shows quantitative analysis of the proportion of cells showing new H3.3 localization at PML-NBs. Numbers represent the mean of 2 independent experiments ± s.d.
PML-NBs are not sites of H3.3 deposition but rather serve as interstages in the chromatin assembly pathway of H3.3
While the precise roles of PML-NBs remains elusive, it was proposed that PML-NBs may serve as sites of storage for proteins, or could be active sites for transcriptional and chromatin regulation.Citation48 Importantly, in our analysis, we observed that both the pools of soluble () and incorporated new H3.3 (histones resistant to detergent extraction, Fig. S10B) localize in PML-NBs, suggesting incorporation of H3.3 into chromatin within the PML-NBs. Given the non-random association of PML-NBs with transcriptionally active regionsCitation62,Citation63 and the importance of the histone variant H3.3 for transcriptional activation,Citation17 we first tested if PML-NBs could be direct sites of H3.3 assembly during transcription. We took advantage of MRC5 cells overexpressing Myc-DAXX, which feature large PML-NBs (as observed in ) that contain DAXX and are surrounded by a thin PML “shell” (). We combined immunostaining of Myc and PML with 5-Ethynyl uridine (EU) to detect localization of the epitope-tagged DAXX proteins within the large PML-NBs and ongoing RNA transcription in vivo. We observed a striking exclusion of de novo RNA synthesis within the shell formed by PML proteins in the large PML-NBs () suggesting that PML-NBs do not contain any detectable nascent RNA. Of note, we also noticed a low DAPI intensity at the place of the large PML-NBs (; Fig. S10A) suggesting absence or low DNA content in these structures, consistent with previous work.Citation64 In addition, we observed that new H3.3 still localizes to PML-NBs upon transcription inhibition with Actinomycin D in proliferating and senescent cells (Fig. S11A and B). Thus, PML-NBs are unlikely to be direct sites of chromatin incorporation for H3.3 and may rather function as storage bodies or indirectly regulate chromatin assembly pathways. Given the physical association of H3.3 with several histone chaperones within PML-NBs (; Fig. S4B), we hypothesized that PML-NBs could be gathering points in the replication-independent chromatin assembly pathway for H3.3 before its deposition in specific genomic locations.
Figure 6. Overexpression of DAXX triggers formation of large PML-NBs together with an accumulation of HP1 proteins in these nuclear bodies. (A) Western blot analysis of total cell extracts from MRC5 cells transduced with an empty vector or a vector expressing Myc-DAXX. Membranes were probed for Myc to verify expression of the transduced proteins, and for DAXX. α-Tubulin is a loading control. M, molecular weight marker. (B) Fluorescent microscopy visualization of PML (green) and DAXX (red) in MRC5 cells treated as in (A) shows formation of large PML-NBs upon Myc-DAXX overexpression (white arrowheads). Scale bar is 10 μm. (C) Fluorescent microscopy visualization of nascent global RNA transcription in vivo in MRC5 cells empty or overexpressing Myc-DAXX. Nascent RNAs were labeled with 5-ethynyl uridine (EU) for 3 h and revealed by the Click-iT chemistry (red). Co-staining with Myc (green) and PML (cyan, pseudo-color) shows absence of transcription within the large PML-NBs upon overexpression of DAXX. Insets represent enlarged images (3×) of selected area. Scale bar is 10 μm. (D and E) Fluorescent microscopy visualization of Myc-DAXX (Myc, green) and HP1γ or H3K9me3 (red) in MRC5 cells treated as in (A) shows accumulation of HP1γ (D) in absence of H3K9me3 (E) in the large PML-NBs upon overexpression of Myc-DAXX (white arrowheads). Scale bar is 10 μm.
H3.3 is targeted to pericentromeric heterochromatin in a DAXX- and PML-dependent manner
To test this hypothesis, we focused on pericentromeric heterochromatin, a typical form of constitutive heterochromatin that is crucial for proper chromosome segregation. It is composed of DNA satellite repeats packed in heterochromatin that is associated with specific histone modifications such as H3K9me3 and is enriched in HP1 proteins.Citation65 Interestingly, H3.3 was recently shown to be enriched in pericentromeric heterochromatin in mouse cellsCitation26 as well as in human cells.Citation66 We first asked if PML-NBs contain pericentromeric heterochromatin markers. Thus, we performed immunofluorescence staining against HP1γ or H3K9me3 in MRC5 cells overexpressing Myc-DAXX. We observed a striking accumulation of HP1γ in the core of the large PML-NBs, but no local increase in H3K9me3 modification (), suggesting the presence of a soluble pool of HP1 proteins within the PML-NBs. Of note, concentration of HP1γ into the large prominent nuclear body was not dependent on transcription or on any RNA component as it persisted in cells treated with Actinomycin D and RNaseA, respectively (data not shown).
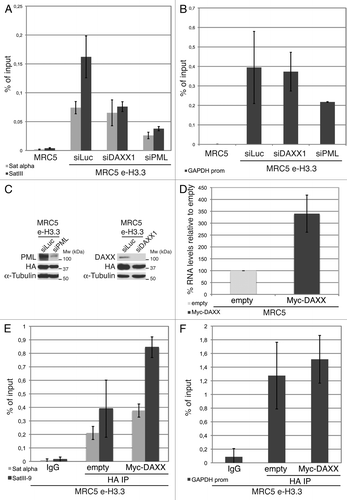
We then used chromatin immunoprecipitation (ChIP) to assess the localization of H3.3 histones in our human primary MRC5 e-H3.3 stable cell lines. In normal MRC5 e-H3.3 cells, H3.3 localizes to the promoter of the active housekeeping gene GAPDH, as well as to centromeric (α-satellite) and pericentromeric heterochromatin regions (satellite III) (). Upon depletion of DAXX by siRNA transfection (), we observed a slight reduction of H3.3 localization at α-satellite regions and a much more prononced reduction at pericentromeric heterochromatin (about 50% reduction) (), whereas H3.3 localization at the GAPDH promoter was unaffected (). Importantly, and in contrast to DAXX depletion, depletion of PML led to a marked decrease in the localization of H3.3 at both centromeric and pericentromeric heterochromatin (), and also at the GAPDH promoter (). These results suggest that depletion of PML (and disruption of PML-NBs) impairs H3.3 deposition at specific genomic locations both in euchromatin and heterochromatin. We next asked if DAXX-mediated targeting of H3.3 to pericentromeric heterochromatin via the PML-NB route would affect the levels of transcription of this region. To this end, we measured the levels of RNA transcripts for the satellite III region by qRT-PCR. In normal MRC5 cells, the level of satellite III transcripts was close to the detection limit, thus preventing us from studying a possible decrease in transcription upon DAXX knockdown. However, upon DAXX overexpression, we observed increased levels of satellite III transcripts as measured by quantitative RT-PCR (). We thus wondered if this increase was associated with an enhanced localization of H3.3 at the satellite III region. We performed ChIP against H3.3 histones in MRC5 e-H3.3 cells overexpressing Myc-DAXX. In comparison with empty MRC5 e-H3.3 cells, H3.3 localization to α-satellite regions was slighly increased, and a pronounced increase was observed at pericentromeric heterochromatin upon DAXX overexpression (). H3.3 localization at the GAPDH promoter remained unaltered (). Together, our results implicate PML-NBs in the histone H3.3 assembly process and support a model in which PML-NBs may play an important role for chromatin organization at specific genomic locations such as pericentromeric heterochromatin.
Figure 7. Enrichment of H3.3 in pericentromeric heterochromatin is dependent on DAXX and PML proteins (A) Histogram shows analysis of H3.3 incorporation in centromeric (satellite α) or pericentromeric heterochromatin (satellite III) by ChIP against the HA epitope in MRC5 e-H3.3 cells treated for 72 h with the indicated siRNAs. QPCR data are presented as fold enrichment of IP over input DNA. MRC5 untransduced cells serve as a negative control. Numbers represent the mean of 2 independent experiments ± s.d. (B) Histogram shows analysis of H3.3 incorporation at the GAPDH promoter region by ChIP assay performed as in (A). Numbers represent the mean of 2 independent experiments ± s.d. (C) Western blot analysis of total cell extracts from MRC5 e-H3.3 cells treated as in (A). Membranes were probed for PML and DAXX to verify depletion of these proteins, and for HA to verify expression of the transduced proteins. α-tubulin is a loading control. M, molecular weight marker. (D) Satellite III mRNA expression levels in empty MRC5 cells or MRC5 cells overexpressing Myc-DAXX as determined by quantitative RT-PCR. mRNA levels were normalized to the reference gene GAPDH and levels were set to 100% in empty cells. Numbers represent the mean of 3 independent experiments ± s.d. (E) Histogram shows analysis of H3.3 incorporation in centromeric (satellite α) or pericentromeric heterochromatin (satellite III) by ChIP against the HA epitope in MRC5 e-H3.3 cells transduced with an empty vector (empty) or a vector encoding Myc-DAXX (Myc-DAXX). QPCR data are presented as fold enrichment of IP over input DNA. IP with IgG control antibody in MRC5 e-H3.3 cells serves as a negative control. Numbers represent the mean of 2 independent experiments ± s.d. (F) Histogram shows analysis of H3.3 incorporation at the GAPDH promoter region by ChIP assay performed as in (E). Numbers represent the mean of 2 independent experiments ± s.d.
Discussion
Our comprehensive analysis of H3.1 and H3.3 deposition in vivo provides novel insights into the dynamics of these H3 variants and their chromatin-assembly pathways in proliferating and senescent primary human cells. We show a global incorporation of new H3.3 in senescent cells with no accumulation into SAHF but instead localization in PML-NBs together with its known chaperones ASF1a, HIRA, DAXX, and ATRX. We further demonstrate a specific role of DAXX in the targeting of H3.3 to PML-NBs in proliferating and senescent cells and we present evidence that this process is important for pericentromeric heterochromatin organization. Together, our results results implicate PML-NBs in the replication-independent chromatin assembly pathway of H3.3.
Replication-independent chromatin assembly as a source of histone replacement in senescent cells
While chromatin structure is inherently dynamic,Citation67 the question of how chromatin is maintained in the absence of DNA replication has remained elusive. In particular, persistance of senescent melanocytes in benign human naevi in the human body for decadesCitation9 highlights the need for chromatin maintenance mechanisms to secure the senescent phenotype. Whereas a global increase in protein content, as well as a specific increase in the macroH2A histone variant have been reported in senescence,Citation44,Citation68 a decrease in the protein levels of the histones H2A, H3, and H4 was described upon drug-evoked senescence or replicative aging,Citation69,Citation70 suggesting that chromatin destabilization is associated with senescence. Here, we extend these results by reporting a dramatic decrease in H3.1 mRNA levels and a mild reduction of H3.3 mRNA levels in OIS (Fig. S3A). In addition, by taking advantage of the SNAP-tagging technology to label new histones in human primary cells, we report that new H3.3 but not H3.1 is incorporated in senescent cells. While this result is not surprising given the downregulation of the H3.1-specific histone chaperone CAF-1 in non-dividing cells,Citation58,Citation71 deposition of H3.3 in senescent cells suggests that replication-independent chromatin assembly is an important mode of histone replacement in non-dividing senescent cells. Recent studies identifying driver H3.3 mutations in pediatric glioblastomas,Citation34-Citation37 or linking H3.3 incorporation with cellular memory,Citation72 further underscore the physiological importance of H3.3 deposition for maintenance of chromatin structure and cell phenotype.
H3.3 dynamics and SAHF formation
Importantly, while formation of SAHF has been associated with the irreversibility of the cell cycle arrest,Citation41,Citation43,Citation73 the molecular basis for their formation has remained elusive.Citation74 SAHF formation is in part regulated by the H3.3 histone chaperones HIRA and ASF1a,Citation44,Citation47 suggesting involvement of the H3.3 histone variant in this process. Here we show that H3.3 does not localize to SAHF during senescence, but is actually excluded from them (; Fig. S3C and D). Rather, H3.3 is found in transcriptionally active regions in senescent cells, consistent with previous data collected in proliferating cells that showed that H3.3 marks actively transcribed genesCitation21-Citation25 and is enriched in post-translational modifications (PTMs) associated with the active chromatin state.Citation75,Citation76 In addition, we did not observe any defects in SAHF formation upon depletion of H3.3 or its chaperones ATRX and DAXX (Figs. S8 and S9), suggesting that these proteins are not required for SAHF formation. This is consistent with recent data showing that SAHF are rather formed through the spatial rearrangement of pre-existing heterochromatin.Citation77 However, absence of H3.3 from SAHF does not preclude its incorporation in other heterochromatic regions such as pericentromeric and telomeric heterochromatin regions (), which localize outside of SAHF.Citation41-Citation43,Citation78 Of note, SAHF are dispensable for cellular senescence and mostly form in response to activated H-Ras in various cell types.Citation79 Thus, deposition of H3.3, which is not linked to SAHF formation, might also be an important process for chromatin maintenance in other types of senescence such as replicative senescence, or drug-induced senescence. Interestingly, we observed an enrichment of pre-existing H3.1 at SAHF ( and ), suggesting that the presence of H3.1 in SAHF may reflect a spatial repositionning of this variant within the H3K9me3-rich core region of SAHF,Citation77 which would be consistent with patterns of repressive PTMs found on H3.1.Citation75,Citation80
PML-NBs are novel regulatory sites for H3.3 replication-independent chromatin assembly
While PML-NBs may have specific functions that are crucial for senescence entry,Citation50,Citation51,Citation81 we show that H3.3 is found in PML-NBs in senescent but also in proliferating cells highlighting a novel role for these nuclear bodies as regulatory sites for H3.3 chromatin assembly pathways. We demonstrate that H3.3 localization to PML-NBs is dependent on DAXX, both in proliferating cells (consistent with data obtained in mouse ES cellsCitation26), and in senescent cells. The structure of DAXX in complex with a dimer of H3.3-H4Citation82,Citation83 suggests that H3.3 may be deposited onto PML-NBs as H3.3-H4 dimers. Indeed, a recent study by Delbarre et al. presents experimental evidence for this.Citation84 In this work, DAXX-dependent H3.3 deposition in PML-NBs was also observed by using transient transfection of epitope-tagged H3.3 in slow growing human cells.Citation84 Using the SNAP technology, we extend these findings to fast proliferating and senescent cells, indicating that localization to PML-NBs may be a universal replication-independent process of H3.3 targeting to chromatin that operates at all times, maybe also in terminally differentiated cells.
Importantly, while H3.3 is found in complex with the other H3.3 chaperonesCitation26,Citation27,Citation33 (), its targeting to PML-NBs does not depend on ATRX and HIRA. For ATRX, a possible explanation is that interaction between ATRX and H3.3 may not be direct, but rather be mediated by DAXX.Citation26 In addition, recruitment of ATRX to PML-NBs is DAXX-dependent (Figs. S5B and S6),Citation60 while the reverse is not true (Fig. S5B). Thus, DAXX seems to be the key factor for the recruitment of ATRX and H3.3 to PML-NBs. Interestingly, our analysis performed in senescent cells revealed that ATRX localizes to SAHF in an increased manner when it is no longer targeted to PML-NBs upon DAXX depletion (Fig. S6). While localization of ATRX at SAHF may be mediated through an interaction with H3K9me3 and HP1α,Citation85,Citation86 it remains to be determined whether this would have an impact on the chromatin dynamics of the macroH2A variant.Citation87
Concerning HIRA and its associated chaperone ASF1a, we hypothesize that their localization in PML-NBs together with H3.3 could be a primary event before targeting of H3.3 to active genes.Citation20,Citation27 Interestingly, PML-NBs associate non-randomly with genomic regions that are transcriptionally active, and nascent RNA has been detected in close proximity to PML-NBs,Citation48,Citation62-Citation64,Citation88 suggesting that PML-NBs could control transcriptional activities indirectly by participating in the replication-independent assembly of H3.3 (). Of note, however, H3.3 loss from PML-NBs upon DAXX depletion does not affect the global incorporation of new H3.3 (; ref. Citation20), making it unlikely that localization of H3.3 to PML-NBs is a way to target this histone variant genome-wide. This also potentially explains the absence of a phenotype for the loss of H3.3 from PML-NBs in regards to senescence (Figs. S8 and S9). Thus, as suggested in Delbarre et al., PML-NBs may be a gathering point for new H3.3 histones before their targeting to specific genomic locations.
Here we present novel evidence to support this hypothesis. First, we show that large PML-NBs are devoid of transcriptional activity () and do not contain large amounts of DNA,Citation64 indicating that PML-NBs are unlikely to be direct sites for incorporation of H3.3 into chromatin and may rather represent novel interstages in the replication-independent pathway. Second, we show that upon DAXX overexpression, formation of large PML-NBs is associated with an accumulation of new H3.3 (; Fig. S10A) together with HP1 proteins, independently of H3K9me3 (), consistent with data obtained in ICF cells.Citation89 These observations strenghten the connection between PML-NBs and heterochromatin componentsCitation89-Citation91 and support a role of these nuclear bodies in heterochromatin dynamics. We hypothesize that the pool of HP1 could serve for pericentromeric heterochromatin organization together with new H3.3. Whether satellite DNA would directly localize in large PML-NBs as observed in G2 phase in ICF cellsCitation89 is currently not known and should be examined further. Third, we show that depletion of DAXX (which abolishes H3.3 localization in PML-NBs) as well as depletion of PML impairs localization of H3.3 at pericentromeric heterochromatin (), and that overexpression of DAXX increases localization of H3.3 at these regions (), thus implicating PML-NBs in heterochromatin organization. Recent data obtained in mouse ES cells showed that PML depletion impairs chromatin structure at telomeres,Citation92 further underscoring the importance of PML-NBs in heterochromatin maintenance.
Interestingly, failure to organize PML-NBs has been associated with the malignant state of prostate cancer,Citation93 and the ATRX-DAXX-H3.3 pathway is mutated in cancers.Citation34-Citation39 How these chromatin assembly pathways are interconnected remains to be investigated. In summary, our data reveal new elements of chromatin maintenance in proliferating and senescent cells and implicate the enigmatic PML-NBs structures in the replication-independent chromatin assembly pathways.
Materials and Methods
Human cell lines and retroviruses
Human MRC5 primary lung fibroblast (ATCC) and human HEK 293T embryonic kidney cells (Intercell, AG) were cultivated in DMEM medium (GIBCO, Life Technologies). Human IMR90 ER:Ras primary lung fibroblast (a kind gift of Dr Narita) were cultivated in DMEM medium without phenol red (GIBCO, Life Technologies). 4-hydroxytamoxifen (4-OHT, SIGMA) was added at a concentration of 100 nM for 6 d to induce senescence. All media contained 10% FCS (GIBCO, Life Technologies), 100 units/mL of penicillin, 100 µg/mL of streptomycin, and 25 µg/mL of Fungizone® (amphotericin B) (Anti-anti, GIBCO, Life Technologies). MRC5 cell lines stably expressing H3.1-SNAP-HAx3 (e-H3.1), H3.3-SNAP-HAx3 (e-H3.3), Myc-hDAXX, or oncogenic Ras (H-RasV12) were established by retroviral transduction.Citation94 Briefly, pBABE plasmids encoding H3.1-SNAP-HAx3 or H3.3-SNAP-HAx3 (gift from Dr Jansen), pLNCX2, pLNCX2 encoding Myc-hDAXX (generated by standard molecular biology procedures), pBABE-puro, pBABE-hygro, pBABE-puro, and pBABE-hygro encoding oncogenic Ras,Citation7 pSuper.retro empty or pSuper.retro shDAXX1Citation95 or shATRX1Citation96 were co-transfected with pCL-ampho plasmidCitation97 by the calcium phosphate method into HEK 293T cells to package retroviral particles.Citation98 After 48 h, supernatant containing replication-incompetent retroviruses was filtered and applied for 24 h on the target MRC5 cells in a medium containing polybrene 8 μg/mL (SIGMA).Citation94 Stable transfectants were selected with Blasticidin S (5 μg/mL, SIGMA), puromycin (1 μg/mL, SIGMA), hygromycin (150 μg/mL, SIGMA), or neomycin (G418, 1 mg/mL, Calbiochem, Millipore) for 3 d, and a polyclonal population of cells was used for all experiments. Even though levels of expression of H3-SNAP-HAx3 were heterogenous within the cell population, we verified the global low expression levels of tagged H3.1 and H3.3 by western blot as compared with the endogenous counterparts.
siRNAs, shRNAs, and transfections
MRC5 cells were transfected in an antibiotics-free medium for the indicated number of days with 40 nM siRNA using Lipofectamine RNAiMax reagent (Invitrogen, Life Technologies) and Opti-MEM 1 medium (GIBCO, Life Technologies) according to manufacturer's instructions. We used the following siRNA sequences: siLuc (non-targeting siRNA against Luciferase): 5′-CGUACGCGGA AUACUUCGA; siDAXX1: 5′-GGAGUUGGAU CUCUCAGAACitation99; siDAXX2: 5′- CAGCCAAGCU CUAUGUCUACitation20; siDAXX3: 5′- CAGAAACAUU AAUAAACAAU ACitation100; siATRX1: 5′-GAGGAAACCU UCAAUUGUACitation96; siATRX2: 5′- GCAGAGAAAU UCCUAAAGACitation96; siH3.3A: 5′-CUACAAAAGC CGCUCGCAACitation101; siH3.3B: 5′-GCUAAGAGAG UCACCAUCACitation101; siHIRA: 5′-GGAUAACACU GUCGUCAUCCitation20; siPML: 5′- GUGCUUCGAG GCACACCAG.Citation102 We used siRNAs against H3.3A and H3.3B together, or ATRX1 and ATRX2 together, at a final concentration of 20 nM each to achieve depletion of H3.3 and ATRX, respectively.
We used the following shRNA sequences cloned into a pSuper.retro vector: shDAXX1: same as siDAXX1,Citation95 shATRX1: 5′-GATCCCCGAG GAAACCTTCA ATTGTATTCA AGAGATACAA TTGAAGGTTT CCTCTTTTTA.Citation96
Antibodies
Table S1 compiles all primary antibodies used in this study. Company, as well as the order number, the lot number, the species, the dilutions for western blotting (WB), and immunofluorescence (IF) are provided for each antibody.
H3-SNAP labeling in vivo
The SNAP-labeling protocol is as described in references Citation52 and Citation54. Four micrometers of SNAP-Block (New England Biolabs) was added during 30 min at 37 °C to the cell medium to quench the SNAP-tag activity or 2 μM of SNAP-Cell TMR-Star (New England Biolabs) during 20 min for pulse labeling. After washing of cells with prewarmed PBS, reincubation in complete medium for 30 min allowed excess compound to diffuse from cells, and then cells were washed again in PBS. For the chase, incubation of cells was for 3 h and 30 min in complete medium at 37 °C. Actinomycin D was added at a dose of 2 μg/mL for the total lenght of the chase (3 h 30 min) to prevent transcription of new H3.3, or at the end of the chase (for 1 h 30 min) to allow synthesis of new H3.3 before transcription inhibition (Fig. S11). After in vivo labeling, the cells were processed for immunostaining.
Immunofluorescence microscopy
Cells grown on coverslips were directly fixed with 4% formaldehyde or pre-extracted with detergent prior to fixation. Indeed, to detect histones incorporated into chromatin only, soluble proteins were extracted for 2 min in cold CSK buffer (10 mM PIPES pH = 7, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2) containing 0.5% of Triton X-100 (SIGMA), and rinsed with CSK and PBS before fixation. For immunofluorescence, we permeabilized cells in PBS containing 0.2% Triton X-100. We then blocked cells with BSA (5% in PBS containing 0.1% Tween 20) for 20 min before incubation with primary antibodies (Table S1) for 1 h, followed by washes in PBS 0.1% Tween, and then incubation with secondary antibodies for 30 min. Cross-absorbed Alexa-488, Alexa-594 or Alexa-647 conjugated secondary antibodies (Invitrogen, Molecular probes) were used to detect primary antibodies. Coverslips were mounted in Vectashield (Vector Laboratories) containing DAPI. To label global nascent RNA transcription in vivo, we incubated cells with 5-Ethynyl uridine (EU, Invitrogen, Molecular probes), a nucleoside analog of uracil incorporated into RNA during active RNA synthesis, at 1 mM for 3 h. Nascent RNA was revealed with the Click-iT Chemistry (Alexa Fluor 594) according to the manufacturer instructions.
Microscopy analysis and quantification of TMR signal
We acquired images with a DMI6000B (Leica) inverted widefield epifluorescence microscope (63× objective/NA 1.32 or 40× objective/NA 1.0) piloted with Leica Application Suite (LAS) and equipped with a Leica DFC365 FX black and white camera or a Leica DFC 295 color camera. We applied identical settings and the same contrast adjustment for all images to allow accurate data comparison, except on , where TMR signal was adjusted to a similar intensity in all conditions to compare localization of new H3.3 at PML-NBs. For brightness and contrast adjustment, we used Adobe Photoshop CS3 (Adobe) and ImageJ.
To quantify fluorescence intensity (TMR signal) in the acquired images, we used the ImageJ software. Briefly, we defined borders of nuclei using HA immunostaining, and we then quantified total gray value for TMR in each nucleus (integrated density). We set a threshold value, above which cells were considered as positive for TMR staining, and then calculated the number of TMR-positive cells for each cell line. We used RGB profiler and interactive 3D surface plot plugins to quantify fluorescence intensity along a line drawn through the nucleus or within all nuclei, respectively. A minimum of 100 nuclei was counted in each experiment.
Western blotting
For total extracts, we lysed cells in Laemmli sample buffer (LSB) 1× (62.5 mM Tris HCl pH = 6.8, 10% glycerol, 2% SDS, 0.002% bromophenol blue and 100 mM DTT). For whole-cell extracts (), after 2 washes in PBS, we lysed cells in lysis buffer (50 mM Tris-HCl, pH = 7.5, 300mM NaCl, 1 mM EDTA, 0.5% NP-40, 5% glycerol) containing protease inhibitors (10 μg/mL leupeptin [SIGMA], 10 μg/mL of pepstatin A [SIGMA], 100 μM PMSF [SIGMA]), and phosphatase inhibitors (5 mM sodium fluoride [SIGMA], 10 mM β-glycerophosphate [SIGMA], 0.2 mM sodium orthovanadate [SIGMA]). We incubated lysates for 20 min on ice and then spinned them for 20 min at 14 000 g. We kept the supernatant as whole-cell extracts. For cellular fractionation (; Fig. S3B), we performed cytosolic and nuclear extracts as described in references Citation80 and Citation103 with buffers containing protease and phosphatase inhibitors as described above. We loaded extracts on SDS-polyacrylamide gels and performed electrophoresis. We used Ponceau 1% (SIGMA) to detect proteins transferred on nitrocellulose membranes. We used primary antibodies as described in Table S1. We used secondary antibodies conjugated with Horseradish peroxidase (HRP) (GE Healthcare Life sciences) and revealed signal by chemiluminescence substrate from Amerscham (SuperSignal West Pico or SuperSignal West Femto) (GE Healthcare Life sciences).
Immunoprecipitation
For immunoprecipitation, we used 150 μg of nuclear extracts from plain MRC5 or MRC5 e-H3.1 and MRC5 e-H3.3 transduced with pBABE-puro or pBABE-puro-H-RasV12 retroviruses for 8 d. We performed the immunoprecipitation for 2 h with HA beads (SIGMA A2095 clone HA-7) in IP buffer (20 mM Tris at pH 7.6, 150 mM NaCl, 3 mM MgCl2, 0.1 mM EDTA, 10% glycerol, 0.1% NP-40) containing protease and phosphatase inhibitors as described above. After 3 washes in IP buffer with a 5 min incubation at 4 °C for each wash, beads were resuspended in LSB 1× buffer and 100 mM DTT, boiled 10 min, and the supernatant was then loaded on an SDS-polyacrylamide gel for further western blot analysis.
Chromatin immunoprecipitation (ChIP)
ChiP was performed as described in reference Citation104. Briefly, MRC5, MRC5 e-H3.1, or e-H3.3 cells, treated or not with siRNAs for 72 h, or transduced with viruses encoding Myc-DAXX for overexpression of DAXX for 7 d were crosslinked in 1% formaldehyde for 10 min at room temperature. After stopping the reaction with 0.125 M of glycine for 5 min at room temperature, cells were washed in PBS, scraped in a tube, and lysed in 85 mM KCl, 5 mM PIPES pH = 8.0, 1% NP-40 containing leupeptin (10 μg/mL), pepstatin (10 μg/mL), and PMSF (1 mM) for 15 min on ice. Cells were homogenized using a glass dounce homogenizer, and nuclei were released with 20 strokes on ice. After centrifugation, nuclei were subsequently lysed in nuclei lysis buffer containing 50 mM Tris-HCl pH = 8.0, 10 mM EDTA, 1% SDS, and protease inhibitors as described above. Sonication was performed using a Bioruptor (Diagenode) for 30 min with 30 s ON-30 s OFF pulses to achieve an average chromatin length of 200–500 bp. After centrifugation, chromatin from 4 × 10e6 cells was diluted 5 times in ChIP dilution buffer (50 mM Tris–HCl pH = 7.4, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholic acid, 1 mM EDTA pH = 8.0 containing protease inhibitors as described above) and immunoprecipitated overnight at 4 °C with 1 μg of Rabbit IgG or HA antibody. Protein A agarose beads (Millipore #16–157) were added for 2 h at 4 °C. After extensive washes as described in reference Citation104, bound chromatin was eluted for 30 min in elution buffer (50 mM NaHCO3, 1% SDS) and decrosslinked overnight at 67 °C in the elution buffer containing 0.54 M NaCl. Samples from input chromatin (10%) were diluted 4 times in elution buffer containing 0.54 M NaCl and decrosslinked as ChIP samples. After treatment of samples with RNaseA for 20 min at 37 °C, DNA was purified with PCR purification kit (QIAGEN). Quantitative PCR was performed as described in Supplementary Materials and Methods.
| Abbreviations: | ||
| ATRX | = | alpha-thalassemia mental retardation X-linked |
| DAXX | = | death-domain associated protein |
| HP1 | = | heterochromatin protein 1 |
| OIS | = | oncogene-induced senescence |
| PML | = | promyelocytic leukemia |
| PML-NBs | = | PML nuclear bodies |
| SAHF | = | senescence associated heterochromatin foci |
Additional material
Download Zip (57.8 MB)Acknowledgments
We thank all members of the departement of gynecology for help and discussion, and especially Dorthe Larsen for fruitful discussions, Geneviève Almouzni for valuable advice and for rabbit ASF1a and PML antibodies, Lars Jansen for the pBABE-H3.1-SNAP-HAx3 and pBABE-H3.3-SNAP-HAx3 plasmids, Fabrizio d'Adda di Fagagna for the pBABE-puro/hygro and pBABE-puro-/hygro-H-RasV12 plasmids, Nathalie Berube for the pSuper.retro.neo shATRX1 and shATRX2, Wade Bresnahan for the pSuper.retro.puro shDAXX, and Masashi Narita for providing the IMR90 ER:Ras cells. Confocal imaging was performed with support from the Center for Microscopy and Image Analysis, University of Zurich. This work was supported by grants from the Swiss National Foundation (project grant 31003A-127450 to MS and Marie-Heim Vögtlin grant PMPDP3_139706 to AC) and by the Kanton of Zürich.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
AC and MS designed research. AC, TO, and MG performed research. DF contributed to research. AC analyzed data. AC and MS wrote the paper.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26988
Related Research Data
References
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25:585 - 621; http://dx.doi.org/10.1016/0014-4827(61)90192-6; PMID: 13905658
- Adams PD. Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene 2007; 397:84 - 93; http://dx.doi.org/10.1016/j.gene.2007.04.020; PMID: 17544228
- Narita M. Cellular senescence and chromatin organisation. Br J Cancer 2007; 96:686 - 91; http://dx.doi.org/10.1038/sj.bjc.6603636; PMID: 17311013
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593 - 602; http://dx.doi.org/10.1016/S0092-8674(00)81902-9; PMID: 9054499
- Ramirez RD, Morales CP, Herbert BS, Rohde JM, Passons C, Shay JW, Wright WE. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev 2001; 15:398 - 403; http://dx.doi.org/10.1101/gad.859201; PMID: 11230148
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou L-VF, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. [Internet] Nature 2006; 444:633 - 7; http://dx.doi.org/10.1038/nature05268; PMID: 17136093
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638 - 42; http://dx.doi.org/10.1038/nature05327; PMID: 17136094
- Mallette FA, Gaumont-Leclerc M-F, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 2007; 21:43 - 8; http://dx.doi.org/10.1101/gad.1487307; PMID: 17210786
- Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436:720 - 4; http://dx.doi.org/10.1038/nature03890; PMID: 16079850
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature 2005; 436:642 - 642; http://dx.doi.org/10.1038/436642a; PMID: 16079833
- Chen Z, Trotman LC, Shaffer D, Lin H-K, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725 - 30; http://dx.doi.org/10.1038/nature03918; PMID: 16079851
- Lazzerini Denchi E, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol 2005; 25:2660 - 72; http://dx.doi.org/10.1128/MCB.25.7.2660-2672.2005; PMID: 15767672
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005; 436:660 - 5; http://dx.doi.org/10.1038/nature03841; PMID: 16079837
- Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair?. Nat Rev Mol Cell Biol 2012; 13:436 - 47; http://dx.doi.org/10.1038/nrm3382; PMID: 22722606
- Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol 2013; 20:14 - 22; http://dx.doi.org/10.1038/nsmb.2461; PMID: 23288364
- Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 2010; 11:264 - 75; http://dx.doi.org/10.1038/nrm2861; PMID: 20197778
- Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res 2011; 21:421 - 34; http://dx.doi.org/10.1038/cr.2011.14; PMID: 21263457
- Wu RS, Tsai S, Bonner WM. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell 1982; 31:367 - 74; http://dx.doi.org/10.1016/0092-8674(82)90130-1; PMID: 7159927
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004; 116:51 - 61; http://dx.doi.org/10.1016/S0092-8674(03)01064-X; PMID: 14718166
- Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, Schultz DC, Pchelintsev NA, Adams PD, Jansen LET, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol Cell 2011; 44:928 - 41; http://dx.doi.org/10.1016/j.molcel.2011.12.006; PMID: 22195966
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 2002; 9:1191 - 200; http://dx.doi.org/10.1016/S1097-2765(02)00542-7; PMID: 12086617
- Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet 2005; 37:1090 - 7; http://dx.doi.org/10.1038/ng1637; PMID: 16155569
- Wirbelauer C, Bell O, Schübeler D. Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev 2005; 19:1761 - 6; http://dx.doi.org/10.1101/gad.347705; PMID: 16077006
- Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet 2009; 41:941 - 5; http://dx.doi.org/10.1038/ng.409; PMID: 19633671
- Chow C-M, Georgiou A, Szutorisz H, Maia e Silva A, Pombo A, Barahona I, Dargelos E, Canzonetta C, Dillon N, Dillon N. Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep 2005; 6:354 - 60; http://dx.doi.org/10.1038/sj.embor.7400366; PMID: 15776021
- Drané P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 2010; 24:1253 - 65; http://dx.doi.org/10.1101/gad.566910; PMID: 20504901
- Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010; 140:678 - 91; http://dx.doi.org/10.1016/j.cell.2010.01.003; PMID: 20211137
- Wong LH, Ren H, Williams E, McGhie J, Ahn S, Sim M, Tam A, Earle E, Anderson MA, Mann J, et al. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res 2009; 19:404 - 14; http://dx.doi.org/10.1101/gr.084947.108; PMID: 19196724
- Ray-Gallet D, Quivy J-P, Scamps C, Martini EM-D, Lipinski M, Almouzni G. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol Cell 2002; 9:1091 - 100; http://dx.doi.org/10.1016/S1097-2765(02)00526-9; PMID: 12049744
- Balaji S, Iyer LM, Aravind L. HPC2 and ubinuclein define a novel family of histone chaperones conserved throughout eukaryotes. Mol Biosyst 2009; 5:269 - 75; http://dx.doi.org/10.1039/b816424j; PMID: 19225618
- Banumathy G, Somaiah N, Zhang R, Tang Y, Hoffmann J, Andrake M, Ceulemans H, Schultz D, Marmorstein R, Adams PD. Human UBN1 is an ortholog of yeast Hpc2p and has an essential role in the HIRA/ASF1a chromatin-remodeling pathway in senescent cells. Mol Cell Biol 2009; 29:758 - 70; http://dx.doi.org/10.1128/MCB.01047-08; PMID: 19029251
- Rai TS, Puri A, McBryan T, Hoffman J, Tang Y, Pchelintsev NA, van Tuyn J, Marmorstein R, Schultz DC, Adams PD. Human CABIN1 is a functional member of the human HIRA/UBN1/ASF1a histone H3.3 chaperone complex. Mol Cell Biol 2011; 31:4107 - 18; http://dx.doi.org/10.1128/MCB.05546-11; PMID: 21807893
- Lewis PW, Elsaesser SJ, Noh K-M, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A 2010; 107:14075 - 80; http://dx.doi.org/10.1073/pnas.1008850107; PMID: 20651253
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tönjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482:226 - 31; http://dx.doi.org/10.1038/nature10833; PMID: 22286061
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al, St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012; 44:251 - 3; http://dx.doi.org/10.1038/ng.1102; PMID: 22286216
- Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012; 22:425 - 37; http://dx.doi.org/10.1016/j.ccr.2012.08.024; PMID: 23079654
- Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, Liu X-Y, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 2012; 124:439 - 47; http://dx.doi.org/10.1007/s00401-012-0998-0; PMID: 22661320
- Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011; 331:1199 - 203; http://dx.doi.org/10.1126/science.1200609; PMID: 21252315
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011; 333:425; http://dx.doi.org/10.1126/science.1207313; PMID: 21719641
- Papamichos-Chronakis M, Peterson CL. Chromatin and the genome integrity network. Nat Rev Genet 2013; 14:62 - 75; http://dx.doi.org/10.1038/nrg3345; PMID: 23247436
- Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113:703 - 16; http://dx.doi.org/10.1016/S0092-8674(03)00401-X; PMID: 12809602
- Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d’Ario G, Montani E, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 2011; 13:292 - 302; http://dx.doi.org/10.1038/ncb2170; PMID: 21336312
- Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006; 126:503 - 14; http://dx.doi.org/10.1016/j.cell.2006.05.052; PMID: 16901784
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 2005; 8:19 - 30; http://dx.doi.org/10.1016/j.devcel.2004.10.019; PMID: 15621527
- Costanzi C, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998; 393:599 - 601; http://dx.doi.org/10.1038/31275; PMID: 9634239
- Rai TS, Adams PD. Lessons from senescence: Chromatin maintenance in non-proliferating cells. BBA - Gene Regulatory Mechanisms 2011; 1–10.
- Ye X, Zerlanko B, Zhang R, Somaiah N, Lipinski M, Salomoni P, Adams PD. Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol Cell Biol 2007; 27:2452 - 65; http://dx.doi.org/10.1128/MCB.01592-06; PMID: 17242198
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 2007; 8:1006 - 16; http://dx.doi.org/10.1038/nrm2277; PMID: 17928811
- Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol 2010; 2:a000661 - 000661; http://dx.doi.org/10.1101/cshperspect.a000661; PMID: 20452955
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000; 406:207 - 10; http://dx.doi.org/10.1038/35021000; PMID: 10910364
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 2000; 14:2015 - 27; PMID: 10950866
- Jansen LET, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 2007; 176:795 - 805; http://dx.doi.org/10.1083/jcb.200701066; PMID: 17339380
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol 2003; 21:86 - 9; http://dx.doi.org/10.1038/nbt765; PMID: 12469133
- Bodor DL, Rodríguez MG, Moreno N, Jansen LET. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr Protoc Cell Biol 2012; Chapter 8:Unit8.8.
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363 - 7; http://dx.doi.org/10.1073/pnas.92.20.9363; PMID: 7568133
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4:1798 - 806; http://dx.doi.org/10.1038/nprot.2009.191; PMID: 20010931
- Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23:798 - 803; http://dx.doi.org/10.1101/gad.519709; PMID: 19279323
- Corpet A, De Koning L, Toedling J, Savignoni A, Berger F, Lemaître C, O’Sullivan RJ, Karlseder J, Barillot E, Asselain B, et al. Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J 2011; 30:480 - 93; http://dx.doi.org/10.1038/emboj.2010.335; PMID: 21179005
- Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A 2003; 100:10635 - 40; http://dx.doi.org/10.1073/pnas.1937626100; PMID: 12953102
- Ishov AM, Vladimirova OV, Maul GG. Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J Cell Sci 2004; 117:3807 - 20; http://dx.doi.org/10.1242/jcs.01230; PMID: 15252119
- Jiang W-Q, Nguyen A, Cao Y, Chang AC-M, Reddel RR. HP1-mediated formation of alternative lengthening of telomeres-associated PML bodies requires HIRA but not ASF1a. PLoS One 2011; 6:e17036; http://dx.doi.org/10.1371/journal.pone.0017036; PMID: 21347226
- Wang J, Shiels C, Sasieni P, Wu PJ, Islam SA, Freemont PS, Sheer D. Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J Cell Biol 2004; 164:515 - 26; http://dx.doi.org/10.1083/jcb.200305142; PMID: 14970191
- Kiesslich A, von Mikecz A, Hemmerich P. Cell cycle-dependent association of PML bodies with sites of active transcription in nuclei of mammalian cells. J Struct Biol 2002; 140:167 - 79; http://dx.doi.org/10.1016/S1047-8477(02)00571-3; PMID: 12490165
- Boisvert F-M, Hendzel MJ, Bazett-Jones DP. Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J Cell Biol 2000; 148:283 - 92; http://dx.doi.org/10.1083/jcb.148.2.283; PMID: 10648561
- Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol 2004; 5:296 - 304; http://dx.doi.org/10.1038/nrm1355; PMID: 15071554
- Morozov VM, Gavrilova EV, Ogryzko VV, Ishov AM. Dualistic function of Daxx at centromeric and pericentromeric heterochromatin in normal and stress conditions. Nucleus 2012; 3:276 - 85; http://dx.doi.org/10.4161/nucl.20180; PMID: 22572957
- Deal RB, Henikoff JG, Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 2010; 328:1161 - 4; http://dx.doi.org/10.1126/science.1186777; PMID: 20508129
- De Cecco M, Jeyapalan J, Zhao X, Tamamori-Adachi M, Sedivy JM. Nuclear protein accumulation in cellular senescence and organismal aging revealed with a novel single-cell resolution fluorescence microscopy assay. Aging (Albany NY) 2011; 3:955 - 67; PMID: 22006542
- Lopez MF, Tollervey J, Krastins B, Garces A, Sarracino D, Prakash A, Vogelsang M, Geesman G, Valderrama A, Jordan IK, et al. Depletion of nuclear histone H2A variants is associated with chronic DNA damage signaling upon drug-evoked senescence of human somatic cells. Aging (Albany NY) 2012; 4:823 - 42; PMID: 23235539
- O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 2010; 17:1218 - 25; http://dx.doi.org/10.1038/nsmb.1897; PMID: 20890289
- Polo SE, Theocharis SE, Klijanienko J, Savignoni A, Asselain B, Vielh P, Almouzni G. Chromatin assembly factor-1, a marker of clinical value to distinguish quiescent from proliferating cells. Cancer Res 2004; 64:2371 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-03-2893; PMID: 15059888
- Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol 2008; 10:102 - 9; http://dx.doi.org/10.1038/ncb1674; PMID: 18066050
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. [Internet] EMBO J 2003; 22:4212 - 22; http://dx.doi.org/10.1093/emboj/cdg417; PMID: 12912919
- Chandra T, Narita M. High-order chromatin structure and the epigenome in SAHFs. Nucleus 2013; 4:23 - 8; http://dx.doi.org/10.4161/nucl.23189; PMID: 23232545
- Hake SB, Garcia BA, Duncan EM, Kauer M, Dellaire G, Shabanowitz J, Bazett-Jones DP, Allis CD, Hunt DF. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J Biol Chem 2006; 281:559 - 68; http://dx.doi.org/10.1074/jbc.M509266200; PMID: 16267050
- McKittrick E, Gafken PR, Ahmad K, Henikoff S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci U S A 2004; 101:1525 - 30; http://dx.doi.org/10.1073/pnas.0308092100; PMID: 14732680
- Chandra T, Kirschner K, Thuret J-Y, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 2012; 47:203 - 14; http://dx.doi.org/10.1016/j.molcel.2012.06.010; PMID: 22795131
- Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 2007; 27:2343 - 58; http://dx.doi.org/10.1128/MCB.02019-06; PMID: 17242207
- Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 2011; 10:457 - 68; http://dx.doi.org/10.4161/cc.10.3.14707; PMID: 21248468
- Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell 2006; 24:309 - 16; http://dx.doi.org/10.1016/j.molcel.2006.08.019; PMID: 17052464
- de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, Lowe SW. PML is a direct p53 target that modulates p53 effector functions. Mol Cell 2004; 13:523 - 35; http://dx.doi.org/10.1016/S1097-2765(04)00062-0; PMID: 14992722
- Elsässer SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature 2012; 491:560 - 5; http://dx.doi.org/10.1038/nature11608; PMID: 23075851
- Liu C-P, Xiong C, Wang M, Yu Z, Yang N, Chen P, Zhang Z, Li G, Xu R-M. Structure of the variant histone H3.3-H4 heterodimer in complex with its chaperone DAXX. Nat Struct Mol Biol 2012; 19:1287 - 92; http://dx.doi.org/10.1038/nsmb.2439; PMID: 23142979
- Delbarre E, Ivanauskiene K, Küntziger T, Collas P. DAXX-dependent supply of soluble (H3.3-H4) dimers to PML bodies pending deposition into chromatin. Genome Res 2013; 23:440 - 51; http://dx.doi.org/10.1101/gr.142703.112; PMID: 23222847
- Dhayalan A, Tamas R, Bock I, Tattermusch A, Dimitrova E, Kudithipudi S, Ragozin S, Jeltsch A. The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum Mol Genet 2011; 20:2195 - 203; http://dx.doi.org/10.1093/hmg/ddr107; PMID: 21421568
- Lechner MS, Schultz DC, Negorev D, Maul GG, Rauscher FJ 3rd. The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem Biophys Res Commun 2005; 331:929 - 37; http://dx.doi.org/10.1016/j.bbrc.2005.04.016; PMID: 15882967
- Ratnakumar K, Duarte LF, LeRoy G, Hasson D, Smeets D, Vardabasso C, Bönisch C, Zeng T, Xiang B, Zhang DY, et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates α-globin expression. Genes Dev 2012; 26:433 - 8; http://dx.doi.org/10.1101/gad.179416.111; PMID: 22391447
- Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays 2004; 26:963 - 77; http://dx.doi.org/10.1002/bies.20089; PMID: 15351967
- Luciani JJ, Depetris D, Usson Y, Metzler-Guillemain C, Mignon-Ravix C, Mitchell MJ, Megarbane A, Sarda P, Sirma H, Moncla A, et al. PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J Cell Sci 2006; 119:2518 - 31; http://dx.doi.org/10.1242/jcs.02965; PMID: 16735446
- Seeler JS, Marchio A, Sitterlin D, Transy C, Dejean A. Interaction of SP100 with HP1 proteins: a link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc Natl Acad Sci U S A 1998; 95:7316 - 21; http://dx.doi.org/10.1073/pnas.95.13.7316; PMID: 9636146
- Kepkay R, Attwood KM, Ziv Y, Shiloh Y, Dellaire G. KAP1 depletion increases PML nuclear body number in concert with ultrastructural changes in chromatin. Cell Cycle 2011; 10:308 - 22; http://dx.doi.org/10.4161/cc.10.2.14551; PMID: 21228624
- Chang FTM, McGhie JD, Chan FL, Tang MC, Anderson MA, Mann JR, Andy Choo KH, Wong LH. PML bodies provide an important platform for the maintenance of telomeric chromatin integrity in embryonic stem cells. Nucleic Acids Res 2013; 41:4447 - 58; http://dx.doi.org/10.1093/nar/gkt114; PMID: 23444137
- Vernier M, Bourdeau V, Gaumont-Leclerc M-F, Moiseeva O, Bégin V, Saad F, Mes-Masson A-M, Ferbeyre G. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev 2011; 25:41 - 50; http://dx.doi.org/10.1101/gad.1975111; PMID: 21205865
- Pear W. Transient transfection methods for preparation of high-titer retroviral supernatants. Curr Protoc Mol Biol 2001; Chapter 9:Unit9.11.
- Cantrell SR, Bresnahan WA. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J Virol 2006; 80:6188 - 91; http://dx.doi.org/10.1128/JVI.02676-05; PMID: 16731959
- Ritchie K, Seah C, Moulin J, Isaac C, Dick F, Bérubé NG. Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol 2008; 180:315 - 24; http://dx.doi.org/10.1083/jcb.200706083; PMID: 18227278
- Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol 1996; 70:5701 - 5; PMID: 8764092
- Sambrook J, Russell DW. Calcium-phosphate-mediated Transfection of Eukaryotic Cells with Plasmid DNAs. CSH Protoc 2006; 2006
- Chen LY, Chen JD. Daxx silencing sensitizes cells to multiple apoptotic pathways. Mol Cell Biol 2003; 23:7108 - 21; http://dx.doi.org/10.1128/MCB.23.20.7108-7121.2003; PMID: 14517282
- Zhang X, Gu L, Li J, Shah N, He J, Yang L, Hu Q, Zhou M. Degradation of MDM2 by the interaction between berberine and DAXX leads to potent apoptosis in MDM2-overexpressing cancer cells. Cancer Res 2010; 70:9895 - 904; http://dx.doi.org/10.1158/0008-5472.CAN-10-1546; PMID: 20935220
- Zhang R, Liu S-T, Chen W, Bonner M, Pehrson J, Yen TJ, Adams PD. HP1 proteins are essential for a dynamic nuclear response that rescues the function of perturbed heterochromatin in primary human cells. Mol Cell Biol 2007; 27:949 - 62; http://dx.doi.org/10.1128/MCB.01639-06; PMID: 17101789
- Perfettini J-L, Nardacci R, Séror C, Bourouba M, Subra F, Gros L, Manic G, Amendola A, Masdehors P, Rosselli F, et al. The tumor suppressor protein PML controls apoptosis induced by the HIV-1 envelope. Cell Death Differ 2009; 16:298 - 311; http://dx.doi.org/10.1038/cdd.2008.158; PMID: 19023333
- Martini E, Roche DM, Marheineke K, Verreault A, Almouzni G. Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. J Cell Biol 1998; 143:563 - 75; http://dx.doi.org/10.1083/jcb.143.3.563; PMID: 9813080
- O’Geen H, Echipare L, Farnham PJ. Using ChIP-Seq Technology to Generate High-Resolution Profiles of Histone Modifications. In: Methods in Molecular Biology. Totowa, NJ: Humana Press; 2011.:265–286.