
microRNAs (miRNAs) are a class of small non-coding RNAs of ~22 nucleotides in length that bind to their target mRNAs (mRNAs) and regulate their expression by translation repression and mRNA degradation. Over the past decade, several miRNA genes have been implicated in human cancers, but their exact function and underlying molecular mechanism in carcinogenesis remain poorly understood. This is a key obstacle in the development of miRNA-based cancer therapeutics. miR-17~92, a cluster of 6 miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92), was the first miRNA gene implicated in cancer. Its overexpression occurs in a broad spectrum of human cancers, including lymphoma, leukemia, and solid tissue cancers. miR-17~92 overexpression is mainly driven by gene amplification at the human chromosome 13q31 region, which harbors the miR-17~92 gene (termed MIR17HG), and by Myc-mediated transcriptional upregulation. A recent RNA sequencing analysis of patient biopsies showed that miR-17~92 expression was upregulated by 2~36-fold in 14 out of 79 (18%) cases of diffuse large B-cell lymphomas and in all the 28 cases of Burkitt lymphomas analyzed. As Myc overexpression is the defining feature of Burkitt lymphoma, these results confirmed that the Myc-miR-17~92 axis is operating in all patients with this malignancy.Citation1
Previous studies showed that miR-17~92 overexpression contributes to carcinogenesis. Thus, retroviral overexpression of miR-17~92 accelerated Myc-driven lymphomagenesis and Notch-driven leukemogenesis,Citation2,Citation3 while a miR-17~92 transgene promoted retinoblastoma initiated by inactivation of the Rb pathway.Citation4 Another study showed that miR-17~92 played important roles in sustaining the optimal growth of Myc-driven lymphoma cell lines in tissue culture and in immunodeficient host.Citation5 However, it was unclear whether elevated miR-17~92 expression, per se, is sufficient to drive carcinogenesis, and exactly what role miR-17~92 plays in Myc-driven carcinogenesis.
A recent study from our lab provided some answers to these important questions.Citation6 First, we created transgenic mice specifically overexpressing miR-17~92 in B cells. Those mice developed B-cell lymphomas with high penetrance, establishing miR-17~92 as a powerful cancer driver and validating its encoded miRNAs and downstream pathways as therapeutic targets (see below). Second, we used CD19-Cre to conditionally delete miR-17~92 in B cells of λ-Myc mice, a commonly used model of Burkitt lymphoma. λ-Myc;CD19-Cre;miR-17~92fl/fl mice exhibited a delay in lymphomagenesis. Strikingly, all lymphomas arising from those mice contained 2 intact miR-17~92 alleles. They escaped CD19-Cre-mediated deletion of miR-17~92 by restricting Myc-driven malignant transformation in CD19-negative early B-cell precursors or in a small fraction of CD19-positive cells with low Cre expression. These results show that Myc-driven lymphomagenesis stringently requires 2 intact alleles of miR-17~92. This, together with a recent study demonstrating that miR-17~92 is essential for the development of retinoblastoma driven by the double deletion of Rb and p53,Citation7 prompts us to hypothesize that the requirement of miR-17~92 is a common denominator of cancers. As miR-17~92 overexpression is found in many human cancers, future studies are warranted to investigate the effect of miR-17~92 deletion on carcinogenesis in other mouse models.
We next explored the molecular mechanisms through which miR-17~92 drives lymphomagenesis. We performed PAR–CLIP analysis of human B cells, identified 868 protein coding genes containing miR-17~92-binding sites that are conserved between human and mouse, and validated a select group of target genes in miR-17~92 transgenic B cells. Our analyses showed that miR-17~92 suppresses the expression of multiple inhibitors of the PI3K (Pten and Phlpp2) and NFκB (Cyld, A20, Itch, Rnf11, and Tax1BP1) pathways, and that both pathways are constitutively active in miR-17~92-driven lymphoma cells. Furthermore, chemical inhibition of either pathway significantly controlled tumor growth and prolonged survival of mice bearing miR-17~92-driven lymphomas. These results suggest that activation of the PI3K and NFκB pathways plays important roles in both the development and maintenance of miR-17~92-driven lymphomas, and that dual targeting of PI3K and NFκB pathways is a potential strategy to treat miR-17~92-driven lymphomas ().
Figure 1. A model for miR-17~92-driven lymphomagenesis and potential therapeutic approaches. The miR-17~92 cluster encodes for 6 distinct miRNAs, which fall into 4 miRNA families (miR-17, miR-18, miR-19, and miR-92 families) based on seed sequence (nucleotides 2–7) homology. miRNAs of the same color have identical seed sequences. Under physiological conditions, miR-17~92 plays an essential role in B-cell development. Gene amplification or Myc-mediated transcriptional upregulation causes miR-17~92 overexpression, which drives lymphomagenesis by inhibiting multiple negative regulators of the PI3K and NFκB pathways. Here we propose 2 approaches (red boxes) to treat miR-17~92-driven lymphomas: specific inhibition of oncogenic member(s) of miR-17~92 and dual targeting of PI3K and NFκB pathways.
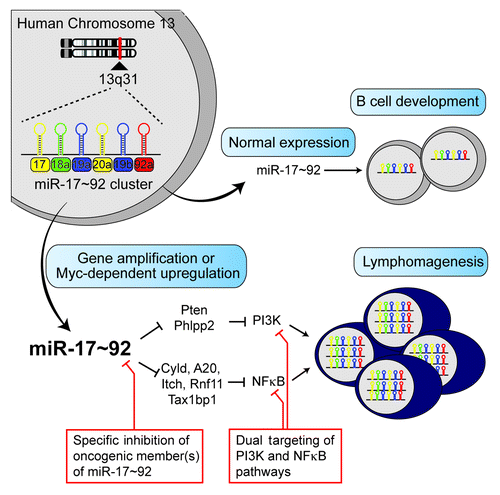
An alternative strategy to treat miR-17~92-driven lymphomas is to sequester or knockdown miR-17~92 miRNAs directly. As miR-17~92 encodes for 6 distinct miRNAs, which may cooperate or even antagonize each other in driving lymphomagenesis, it will be essential to identify the oncogenic component(s) of this cluster before exploring ways to target them. Previous studies identified miR-19 as the sole oncogenic miRNA in the miR-17~92 cluster in the context of cooperating with Myc and Notch,Citation3,Citation8 as well as in sustaining the optimal survival of Myc-driven lymphoma cell lines.Citation5 Nevertheless, it remains unclear whether overexpression of miR-19 alone is sufficient to drive lymphomagenesis, or overexpression of more than one miRNA in the cluster is necessary to initiate malignant transformation. It is equally unclear whether miR-17~92-driven lymphomas are addicted to overexpression of the whole cluster, or addicted to some miRNAs of the cluster, or not addicted to them at all. The answers to these questions will determine whether targeting miR-17~92 miRNAs is a valid strategy to treat miR-17~92-driven lymphomas and, if so, which miRNAs are the right ones to target ().
References
- Schmitz R, et al. Nature 2012; 490:116 - 20; http://dx.doi.org/10.1038/nature11378; PMID: 22885699
- He L, et al. Nature 2005; 435:828 - 33; http://dx.doi.org/10.1038/nature03552; PMID: 15944707
- Mavrakis KJ, et al. Nat Cell Biol 2010; 12:372 - 9; http://dx.doi.org/10.1038/ncb2037; PMID: 20190740
- Conkrite K, et al. Genes Dev 2011; 25:1734 - 45; http://dx.doi.org/10.1101/gad.17027411; PMID: 21816922
- Mu P, et al. Genes Dev 2009; 23:2806 - 11; http://dx.doi.org/10.1101/gad.1872909; PMID: 20008931
- Jin HY, et al. EMBO J 2013; 32:2377 - 91; http://dx.doi.org/10.1038/emboj.2013.178; PMID: 23921550
- Nittner D, et al. Nat Cell Biol 2012; 14:958 - 65; http://dx.doi.org/10.1038/ncb2556; PMID: 22864477
- Olive V, et al. Genes Dev 2009; 23:2839 - 49; http://dx.doi.org/10.1101/gad.1861409; PMID: 20008935