
Keywords: :
Oncogenic stress induces the transcription of the tumor suppressor ARF, which regulates cellular responses predominantly through p53 activation. Specifically, ARF inhibits the degradation of p53 by blocking either MDM2 or ARF-BP1, known E3 ubiquitin ligases for p53 (reviewed in ref. Citation1). Once activated, p53 initiates cell cycle arrest or apoptosis functioning as effective barriers against tumorigenesis. As the transcriptional activation of ARF is well studied upon various cellular stress, recent focus has turned to post-translational regulation of ARF. Interestingly, ARF is a very stable protein in cancer cells yet short-lived in many normal human cell lines. ARF predominantly localizes in the nucleolus and complexes with nucleophosmin (NPM), which helps preserve ARF stability and heighten its function. Accordingly, our group previously aimed to identify potential interacting proteins with ARF through biochemical purification of NPM followed by mass spectrometry. We identified a novel E3 ubiquintin ligase for ARF, coining it ubiquitin ligase for ARF or ULF.Citation2 Inhibition of ULF increases ARF levels, thereby activating p53 and decreasing cellular proliferation. Further, we show that NPM prevents ULF-mediated ubiquitination of ARF by sequestering ARF in the nucleolus and not the nucleoplasm where ULF localizes.Citation2 Lastly, as c-MYC is a potent activator of ARF transcription, we analyzed the interaction between ULF and ARF. Indeed, expression of c-MYC neutralizes ULF-mediated ARF ubiquitination and stabilizes ARF protein levels.Citation2
Aberrant expression of c-MYC is a common driver to many cancers; high levels of c-MYC activate the ARF/p53 axis, which inhibits cell proliferation, while low levels seem to circumvent ARF activation and promote tumorigenesis. Interestingly, basal levels of c-MYC are required for cellular homeostasis and development. Therefore, we aimed to mechanistically dissect the paradoxical roles of the cellular response difference between high and low c-MYC levels in terms of ARF regulation. Infecting cells with c-MYC at 2 different multiplicities of infection (moi), designated as either high or low c-MYC, Chen et al. show that cells infected under high c-MYC levels grew significantly slower than cells infected under low c-MYC conditions. Both conditions activated ARF transcription, yet only high c-MYC infection resulted in ARF protein elevation and subsequent p53-mediated cellular responses.Citation3 High c-MYC infection disrupted the ULF-mediated ARF ubiquitination, allowing for ARF stabilization and p53-dependent apoptosis, which was exacerbated when combined with genotoxic stress. Interestingly, ARF expression gradually decreased after DNA damage, while its mRNA remained stable. Combining DNA damage with ULF knockdown rescued both the ARF depletion and starkly increased p53-dependent apoptosis.Citation3 Taken in aggregate, disrupting ULF-mediated ubiquitination of ARF in combination with either oncogenic and/or genotoxic stress stabilizes ARF and primes cells for p53-mediated apoptosis or cell cycle arrest (). These results, in part, help understand how cells with basal levels of c-MYC do not activate the ARF/p53 axis.
Figure 1. Schematic of ARF protein stability upon oncogenic or genotoxic stress. High levels of c-MYC can prevent ULF-mediated ARF degradation. Upon DNA damage, ATM activates the phosphatase PP1 to dephosphorylate and inactivate NPM, allowing for ARF degradation. Upon ATM depletion, NEK2 can phosphorylate NPM, which increases binding to ARF, allowing subsequent p53 activation to prevent tumorigenesis. Ub, ubiquitination; p, phosphorylation.
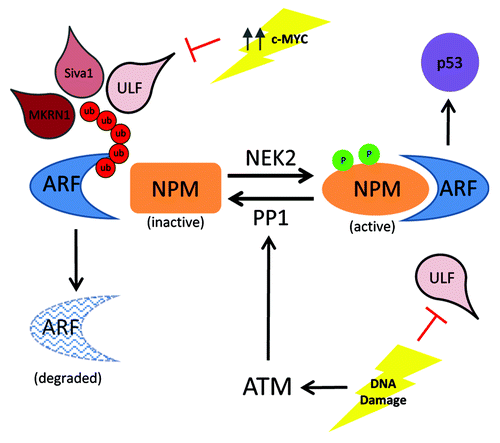
The interplay between DNA damage and ARF stability warrants more study, as ubiquitin-mediated ARF degradation may not be the sole regulator of ARF protein levels. Recently, Vassilis Gorgoulis’ group defined new players in the pathway regulating ARF protein stability.Citation4 Previously reported, yet through unknown mechanisms, mouse embryonic fibroblasts null for ATM, a protein kinase that regulates the DNA damage response cascade, have elevated ARF proteins levels.Citation5 Velimezi et al. extended this observation to human cancer cells through ATM ablation. ARF transcription is unaffected upon ATM depletion, yet ARF protein levels are stabilized. Conversely, activation of ATM through DNA damage decreased ARF protein levels, which was reversed by ATM abrogation. Interestingly, concomitant depletion of NPM and ATM did not result in ARF stabilization, suggesting that ATM regulates NPM function. Surprisingly, by inhibiting ATM, NPM phosphorylation increased (showing stronger binding/sequestration ability for ARF), while under DNA damage, NPM phosphorylation decreased (having weaker binding to ARF).Citation4 This suggested that ATM activates a phosphatase to target NPM. PP1 is a reported ATM-activated phosphatase that also dephosphorylates NPM. Indeed, after PP1 depletion, ARF levels increased and remained high with genotoxic stress, as phosphorylated NPM blocks ARF degradation.Citation4 Next, seeking out the kinase involved in phosphorylating NPM upon genotoxic stress, researchers intuitively chose NEK2, a kinase previously reported to interact with NPM. Dual inhibition of NEK2 and ATM prevented ARF upregulation. Collectively, these experiments show novel opposing roles of PP1 and NEK2 in the ATM–ARF pathway ().Citation4 Lastly, Velimezi et al. showed upon ATM depletion, cells retaining ARF underwent an ARF-dependent cell cycle arrest. This was further appreciated in vivo, when cancer cells retaining ARF function were inhibited for ATM, which led to ARF upregulation and a subsequent decrease in tumorigenesis, while concomitant suppression of ATM and ARF exacerbated tumor growth.Citation4
Preventing ARF degradation in tumors that retain both ARF and p53 function is an attractive therapeutic option. These recent findings from Chen et al. and Velimezi et al. extend the options to combat tumorigenesis through either targeting the regulators of NPM phosphorylation by inhibiting PP1 or activating NEK2, which will block ARF turnover or directly attempt to block ULF. Further, 2 additional E3 ligases targeting ARF have been recently identified: Siva1Citation6 and MKRN1 ().Citation7 How different stresses regulate the interaction of these new E3 ligases and ARF stability has yet to be determined, yet the potential to combat tumorigenesis through ARF stabilization and p53 activation seems promising and warrants more study.
References
- Sherr CJ. Nat Rev Cancer 2006; 6:663 - 73; http://dx.doi.org/10.1038/nrc1954; PMID: 16915296
- Chen D, et al. Nature 2010; 464:624 - 7; http://dx.doi.org/10.1038/nature08820; PMID: 20208519
- Chen D, et al. Mol Cell 2013; 51:46 - 56; http://dx.doi.org/10.1016/j.molcel.2013.05.006; PMID: 23747016
- Velimezi G, et al. Nat Cell Biol 2013; 15:967 - 77; http://dx.doi.org/10.1038/ncb2795; PMID: 23851489
- Kamijo T, et al. Cancer Res 1999; 59:2464 - 9; PMID: 10344759
- Wang X, et al. Nat Commun 2013; 4:1551; http://dx.doi.org/10.1038/ncomms2533; PMID: 23462994
- Ko A, et al. J Natl Cancer Inst 2012; 104:1660 - 72; http://dx.doi.org/10.1093/jnci/djs424; PMID: 23104211