
Abstract
Sister chromatid separation creates a sudden loss of tension on kinetochores, which could, in principle, re-activate the spindle checkpoint in anaphase. This so-called “anaphase problem” is probably avoided by timely inactivation of cyclin B1-Cdk1, which may prevent the spindle tension sensing Aurora B kinase from destabilizing kinetochore–microtubule interactions as they lose tension in anaphase. However, exactly how spindle checkpoint re-activation is prevented remains unclear.
Here, we investigated how different degrees of cyclin B1 stabilization affected the spindle checkpoint in metaphase and anaphase. Cells expressing a strongly stabilized (R42A) mutant of cyclin B1 degraded APC/CCdc20 substrates normally, showing that checkpoint release was not inhibited by high cyclin B1-Cdk1 activity. However, after this initial wave of APC/CCdc20 activity, the spindle checkpoint returned in cells with uncohesed sister chromatids. Expression of a lysine mutant of cyclin B1 that is degraded only slightly inefficiently allowed a normal metaphase-to-anaphase transition. Strikingly, however, the spindle checkpoint returned in cells that had not degraded the cyclin B1 mutant 10–15 min after anaphase onset. When cyclin B1 remained in late anaphase, cytokinesis stalled, and translocation of INCENP from separated sister chromatids to the spindle midzone was blocked. This late anaphase arrest required the activity of Aurora B and Mps1. In conclusion, our results reveal that complete removal of cyclin B1 is essential to prevent the return of the spindle checkpoint following sister chromatid disjunction. Speculatively, increasing activity of APC/CCdc20 in late anaphase helps to keep cyclin B1 levels low.
Introduction
Faithful division of the genome during mitosis is supported by the spindle checkpoint which creates the opportunity for paired sister chromatids to attach bipolarly to the mitotic spindle. The spindle checkpoint represses APC/CCdc20 during prometaphase. This stabilizes cyclin B1, maintaining Cdk1 activity and keeping cells in mitosis, and Securin, which safeguards the cohesion of sister chromatids. In metaphase, APC/CCdc20 becomes highly active, supporting the degradation of cyclin B1 and thereby the inactivation of Cdk1. This initiates spindle elongation and cytokinesis. With remarkable synchrony, APC/CCdc20 -dependent degradation of Securin liberates Separase, the protease that cuts the Cohesin rings, so that sister chromatids are separated and anaphase begins.Citation1,Citation2 Although there are examples of cross-talk between these two events, e.g., Separase supports cytokinesis in yeast and cyclin B1 can influence Separase activity,Citation3-Citation5 there is also evidence that sister chromatid separation occurs entirely independently of cyclin B1 degradation.Citation2,Citation6-Citation8
Coordination between anaphase and mitotic exit is therefore mostly dependent on the spindle checkpoint, which determines the time when both Securin and cyclin B1 start to be degraded. Subsequently, synchrony in the progression of anaphase and cytokinesis is tuned by the efficiencies by which Cohesin is cleaved and phosphorylation events downstream of cyclin B1-Cdk1 are reverted. Activating Separase too slowly, for instance as a result of non-degradable Securin expression, delays sister chromatid separation until after cytokinesis and causes a cut-phenotype.Citation2 In return, failing to degrade cyclin B1 on time will block cell division even though sister chromatids may separate. However, the extent to which cyclin B1 degradation affects mitotic exit remains unclear. Different effects of stabilized cyclin B1 mutants on metaphase, anaphase and cytokinesis have been reported.Citation2,Citation7
In prometaphase, an active spindle checkpoint functions by creating enough time to destabilize erroneous kinetochore-spindle attachments. This allows the paired kinetochores to capture microtubules from opposite poles and let them form correct, stable bipolar attachments. Only several minutes later however, when cells reach anaphase, an active spindle checkpoint would become problematic, by repressing APC/CCdc20 activity that is required for sister chromatid segregation and cytokinesis. So, although in early mitosis cells should detect the liberation of sister chromatids as a reduction of the pulling forces on kinetochores, at anaphase this detection mechanism should be inactivated. It is quite imaginable that failing to control this so-called anaphase problemCitation9 could lead to aneuploidy, such as occurs in cancer.
The problem appears to be avoided by the timely inactivation of Cdk1 prior to anaphase, in metaphase.Citation10-Citation15 It is unknown at the moment whether the anaphase problem could occur naturally, or may contribute to aneuploidy in cases when Cdk1 regulatory factors are abnormally expressed. How inactivation of Cdk1 prevents the anaphase problem is also still largely unclear. It seems logic that repressing the spindle tension-sensing kinase Aurora B around the metaphase-to-anaphase transition would prevent the checkpoint from coming back.Citation11 In line with this, there is a potential role for translocation of INCENP away from sister chromatids after metaphase, as a result of INCENP dephosphorylation. This may at least partially prevent Aurora B kinase from destabilizing kinetochore-spindle attachments.Citation11,Citation14,Citation15 However, Aurora B translocation is clearly not the only mechanism involved, because forcing Aurora B to stay bound to anaphase centromeres per se is insufficient to trigger checkpoint re-activation.Citation11 This means that, perhaps, catalytic activity of Aurora B is downregulated specifically on separating sister chromatids or that yet unknown factors are involved in preventing the checkpoint from returning in late mitosis.
Here, we investigated the hypothesis that subtly reducing the efficiency by which cyclin B1 disappears, thereby shifting the timing of Cdk1 inactivation roughly from metaphase to anaphase, could already be sufficient to re-activate the spindle checkpoint. Indeed, we find that the spindle checkpoint returns within 10–15 min after sister chromatid disjunction, in case residual cyclin B1 remains at this time. This quickly stalls mitotic exit. The presence of cyclin B1 in late anaphase also prevents full translocation of INCENP from separated sister chromatids. The late mitotic arrest resulting from spindle checkpoint re-engagement can be overcome by inhibiting the bipolar-attachment sensing kinase Aurora B or the spindle checkpoint kinase Mps1. To our surprise, we found that poorly degradable mutants of cyclin B1 turned into more efficient APC/C substrates upon checkpoint inhibition in anaphase-arrested cells. This suggest that APC/CCdc20 becomes more active during anaphase, compared with its metaphase activity right after spindle checkpoint release. Indeed, Cdc20 appears to play a role in anaphase activity of the APC/C.Citation16 Alternatively, Aurora B or Mps1 kinase inhibitors release some activity of the other APC/C activator, Cdh1, even though cyclin B1-Cdk1 is still active. The latter possibility would imply that APC/CCdh1 recognizes poor APC/C substrates (such as cyclin B1 with a point-mutation in its D-box, an APC/C substrate which furthermore has no obvious D-box or KEN box) better than APC/CCdc20 does. Summarized, our results reveal that to prevent mitotic failure and thus to safeguard genomic stability, cells must ensure that all cyclin B1 is cleared right after the sister chromatids separated, at its latest.
Results and Discussion
APC/CCdc20 activation and anaphase onset in cells expressing non-degradable cyclin B1
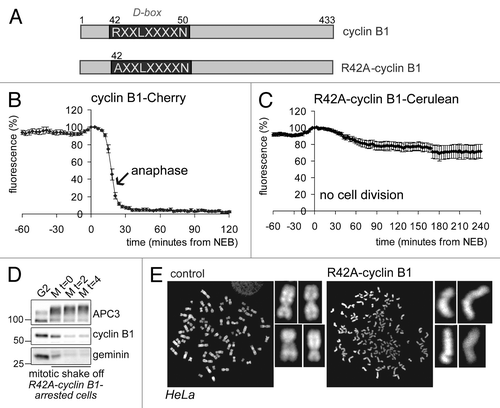
To study the role of cyclin B1 destruction in spindle checkpoint inactivation, we first followed mitotic progression of cells expressing a fluorescently tagged cyclin B1, point-mutated in the destruction box (D-box, R42A; ), which was largely stabilized in mitosis (compare ). In line with our previous observations, these cells arrested completely in mitosis, although they fully degraded their endogenous spindle-checkpoint sensitive APC/CCdc20-substrates ( shows degradation of endogenous cyclin B1 and geminin in cells arrested in mitosis by a fluorescently tagged R42A-cyclin B1; for experiments showing Securin degradation under similar conditions, and further controls, see refs. Citation8 and Citation17). Importantly, sister chromatids became fully separated during the arrest, indicating that the failure to exit mitosis did not hamper the completion of anaphase (). The arrest invoked by non-degradable cyclin B1 has been referred to as a “pseudometaphase” state,Citation7 although our data and results published by others show that it resembles more closely a post-metaphase, anaphase-like state.Citation2,Citation8,Citation17 We conclude that, at least initially, the spindle checkpoint is normally inactivated when cells reach metaphase even though cells express elevated levels of cyclin B1 and cannot exit mitosis.
Figure 1. Cells expressing non-degradable cyclin B1 arrest in mitosis with separated sister chromatids. (A) Schematic overview of the cyclin B1 mutant used. The destruction-box (D-box) is indicated; (B) The fluorescence intensity in live U2OS cells expressing wt-cyclin B1-Cherry was plotted against time in mitosis from NEB. The arrow indicates the start of sister chromatid separation; (C) The fluorescence intensity in live U2OS cells expressing R42A-cyclin B1-Cerulean was plotted against time from NEB. These cells arrest in a post-metaphase state; (D) U2OS cells were transfected with R42A-cyclin B1-Cerulean, thymidine synchronized and released for 14 h. After mitotic shake-off, cells were released in fresh medium for the indicated time in hours. APC/C substrates geminin and cyclin B1 were normally degraded during the arrest. APC3 is used as a loading control; (E) U2OS cells were transfected with R42A-cyclin B1-Venus and collected and processed for chromosome spreads 46 h later. Chromosomes were stained with DAPI. Note that cells expressing R42A-cyclin B1-Venus fully separated their sister chromatids, while all the chromosomes are cohesed in control cells.
It has been shown that Cdk1 can have both positive and negative effects on mitotic exit, for instance, by activating APC/CCdc20 through phosphorylation, as well as by inhibiting APC/CCdc20 by maintaining the spindle checkpoint.Citation12,Citation18,Citation19 Therefore, subsequently we asked to which degree cyclin B1 must be removed in order to prevent spindle checkpoint re-activation in anaphase. We filmed cells expressing a new cyclin B1 mutant, Δ52–85-cyclin B1, which has a deletion within a lysine-rich region that we noticed to be conserved downstream of the D-box (; Fig. S1A shows the mutant binds normally to Cdk1). This region may be needed for efficient ubiquitination by APC/CCdc20 and the E2 enzymes Ube2S and UbcH10.Citation20,Citation21 Interestingly, when followed through mitosis, cells expressing Δ52–85-cyclin B1 appeared to undergo either one of two possible fates: cells arrested after they first started to exit mitosis, in late anaphase, and then realigned their chromosomes and withdrew the incoming cleavage furrow (, red arrows; Fig. S1B), or they divided slightly slowly but otherwise normally and successfully produced two daughter cells (; yellow arrows; also see below; and ).
Figure 2. A lysine mutant of cyclin B1 that is degraded slightly less efficiently can halt mitotic exit after the metaphase-to-anaphase transition. (A) Schematic overview of cyclin B1 mutants. The position of the Lysine-rich region is indicated; (B) Cells that arrest after transfection with Δ52–85-cyclin B1-Cerulean first progress from metaphase to anaphase, before getting stuck in a late anaphase arrest. In the cell indicated with the red arrow, sister chromatid separation is indicated, after which the cell arrests, the cleavage furrow regresses and the separated sister chromatids move back to the equatorial plane of the cell. The cell indicated with the yellow arrow, however, manages to degrade the cyclin B1 mutant and subsequently divides normally. Roughly 50% of the cells expressing this Lysine mutant of cyclin B1 arrest in anaphase.
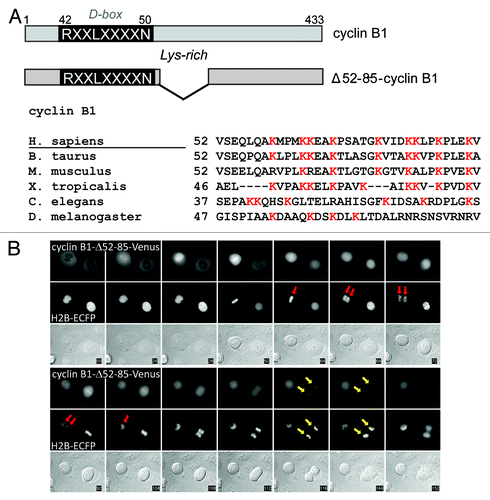
Figure 3. Inefficient degradation of cyclin B1 in anaphase re-activates the spindle checkpoint right after sister chromatid disjunction. The fluorescence intensity in live U2OS cells expressing Δ52–85-cyclin B1-Cerulean was plotted against time from NEB; (A) cells that divided normally. The arrow indicates the start of sister separation. (B) cells that arrested in a post-metaphase state; (C) Bar graph of the fluorescence levels at start of sister separation of cells plotted in (A and B); (D) Comparison of the fluorescence levels at the start of sister chromatid separation as compared with 12 min after sister chromatid separation in cells that either divided (plotted in A) or arrested (plotted in B).
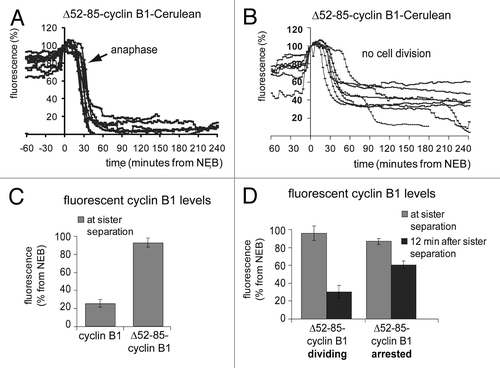
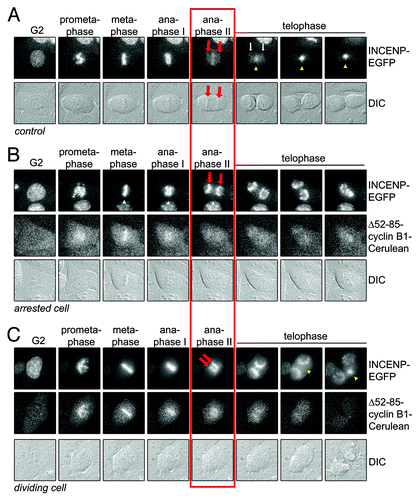
Figure 4. INCENP translocation away from sister chromatids is blocked by residual cyclin B1 at anaphase. (A) Live U2OS cells expressing INCENP-EGFP, as a marker for the CPC, were imaged by DIC and fluorescence microscopy. Still images of the indicated phases in mitosis are shown. Red arrows point to the separating sister chromatids. Yellow arrowheads point to the location where the midzone is formed; (B) U2OS cell co-expressing INCENP-EGFP and Δ52–85-cyclin B1-Cerulean arresting in anaphase. Cells were imaged by DIC and fluorescence microscopy. Still images of the indicated phases in mitosis are shown. Red arrows point to the separating sister chromatids. No midzone is formed and INCENP-EGFP translocation is completely prevented; (C) Example of a cell that divides into two. Red arrows point to the separating sister chromatids. Yellow arrowheads point to the location where the midzone is formed. INCENP-EGFP translocation takes p[lace but is delayed compared with control cells.
We then plotted the cyclin B1 degradation kinetics of these two classes of cells separately. In cells that managed to divide successfully, degradation of Δ52–85-cyclin B1-Cerulean was complete about 15 min after anaphase onset (). In arrested cells however, Δ52–85-cyclin B1 started to be degraded normally, but suddenly became fully re-stabilized when fluorescence levels reached approximately 40% of its starting levels (). The remaining levels of ectopic cyclin B1 at the time of sister chromatid separation were compared with the levels 12 min later and plotted in bar graphs (). First, this showed that cyclin B1 had disappeared with faster kinetics as compared with the lysine-mutant Δ52–85-cyclin B1 (). Additionally, cells in which the efficiency of Δ52–85-cyclin B1 degradation during anaphase was low, arrested in anaphase, whereas cells that degraded the mutant cyclin B1 protein slightly more efficiently, divided (, 12 min after sister separation, compare Δ52–85-cyclin B1-arresting, 51 ± 3,3% and Δ52–85-cyclin B1-dividing, 25 ± 6%; mean ± SEM). The difference in cyclin B1 levels remaining between dividing and arresting cells became apparent only after the start of anaphase, so by the time the separated chromatids started to move toward opposite poles. Altogether, these results indicate that when cyclin B1 is degraded inefficiently and low levels remain right after anaphase, the spindle checkpoint might come back and revert the progression of mitotic exit.
CPC midzone translocation is impaired by Δ52–85-cyclin B1 expression
Removing the tension-sensor Aurora B kinase from centromeres may help to prevent re-activation of the spindle checkpoint in anaphase.Citation10,Citation14,Citation15 Here, we measured the translocation of a fluorescently-tagged version of the Aurora B-binding chromosomal passenger protein INCENP. In control cells, all INCENP-EGFP translocated from the DNA toward the midzone 10–15 min after sister chromatid separation (). However, in cells that arrested in anaphase with elevated levels of cyclin B1, translocation of INCENP-EGFP to the midzone was blocked completely (). Cells that did manage to divide despite expressing Δ52–85-cyclin B1 showed a slightly intermediate level of INCENP-translocation to the midzone, which was delayed until telophase and remained partially incomplete (). Our data indicate that INCENP translocation is already prevented by slightly increased levels of cyclin B1 in anaphase. They also show that the efficiency of INCENP translocation correlates with preventing the anaphase problem.
Inefficient cyclin B1 destruction re-activates the spindle checkpoint
Subsequently, we investigated in more detail whether spindle checkpoint signaling returns in cells that expressed stabilized forms of cyclin B1. To investigate the entire cellular activity of the spindle checkpoint, we analyzed the amount of checkpoint proteins bound to APC/CCdc20-complexes in purified fractions of mitotic cells. Cells expressing R42A-cyclin B1, or Δ52–85-cyclin B1, that delayed in mitosis were specifically collected by mitotic shake-off. These purified mitotic cells arrested by non-degradable cyclin B1 were compared with untransfected mitotic cells released from a nocodazole-block in the presence of proteasome inhibitor MG132 (). In the latter cells, the SAC should not become re-activated since they arrest with high levels of Securin and do not separate their sister chromatids. APC/C immunoprecipitations (; anti-APC4 IP) show a reduction of APC/C-bound spindle checkpoint proteins BubR1 and Mad2 after release from nocodazole into MG132. However, the lysates from cells expressing stabilized cyclin B1 mutants, which as we showed above arrest after sister chromatid separation, contained APC/CCdc20 that remained bound to Mad2, and, to a lesser extent, to BubR1 (; third and fourth lanes of the APC4 IPs). To confirm that the late anaphase arrest was dependent on re-activation of the spindle checkpoint, cells arrested in late anaphase by Δ52–85-cyclin B1-Cerulean were then treated with Aurora B inhibitor AZD1152. This inactivates the spindle checkpoint when kinetochores attach to the spindle but are no longer under tension, such as would happen in anaphase. We measured degradation of the cyclin B1 mutant as a read-out for checkpoint inactivation (; but also see below; ). Indeed, the spindle checkpoint was inactivated by AZD1152 and, after drug addition, cells rapidly degraded the cyclin B1 mutant that had been re-stabilized in anaphase ().
Figure 5. Residual cyclin B1 during anaphase re-activates the spindle checkpoint. (A) U2OS cells were transfected with R42A-cyclin B1-Cerulean or Δ52–85-cyclin B1-Cerulean, thymidine synchronized, released for 14 h and then collected by mitotic shake-off. Lysates from mitotic cells were subjected to APC4-IPs to immunoprecipitate APC/C and analyze co-immunoprecipitation of mitotic checkpoint proteins by western blot. Note that checkpoint proteins BubR1 and Mad2 are removed from the APC/C after release from Nocodazole, but stay bound to the APC/C in mitotic cells expressing cyclin B1-mutants; (B) Fluorescence plot of Δ52–85-cyclin B1-Cerulean in an anaphase arrested cell before and after the addition of Aurora B inhibitor AZD-1152, which inactivates the spindle checkpoint when all kinetochores are attached to the mitotic spindle. The cyclin B1 mutant is is stabilized during the mitotic arrest that it invokes but becomes destabilized again by the addition of the Aurora B inhibitor.
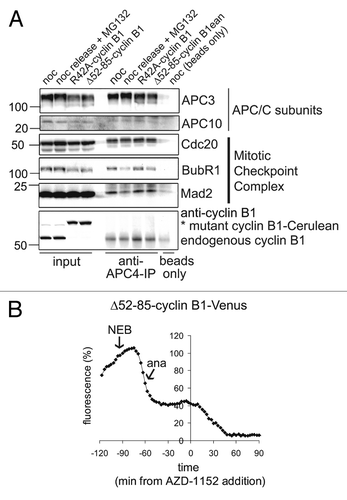
Figure 6. Checkpoint override or Aurora B inhibition broadens the substrate specificity of the APC/C in anaphase. (A) U2OS cells, as in , that were arrested in anaphase by Δ52–85-cyclin B1-Venus, were treated with the Aurora B inhibitor AZD-1152; mitotic progression was monitored by time-lapse microscopy; (B) U2OS cells co-expressing D-box mutated R42A-cyclin B1-Venus, and Histone 2B-ECFP, were followed as they progressed through mitosis after addition of the Aurora B inhibitor AZD-1152; (C) U2OS cells were transfected with R42A-cyclin B1-Cerulean, Δ52–85-cyclin B1-Cerulean or ΔN-cyclin B1-Cerulean, thymidine synchronized, released for 14 h and collected by mitotic shake-off. These mitotic cells were then treated with the Mps1 inhibitor Reversine, for 2 h, and collected for western blots. Mad2 is used as loading control. Cells arrested in mitosis by R42A-cyclin B1, or Δ52–85-cyclin B1, exited mitosis after the addition of Mps1 inhibitor Reversine, as apparent from APC3 de-phosphorylation. However, cells arrested in mitosis by ΔN-cyclin B1, a mutant that cannot be detected by Cdc20 at all, stay arrested in mitosis even after the spindle checkpoint was inactivated by Reversine.
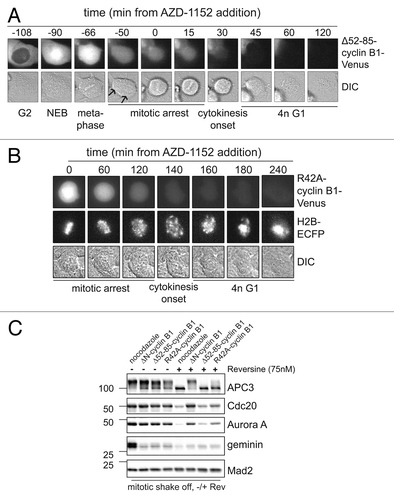
We were surprised that the partially stabilized Δ52–85-cyclin B1-Cerulean lysine mutant, which we suspect is a poor APC/CCdc20 substrate, and thereby re-activates the spindle checkpoint in anaphase, became a better APC/C substrate as soon as we added an Aurora B kinase inhibitor. These cells rapidly progressed through anaphase (). They did not complete cytokinesis, probably due to inhibition of Aurora B which is required for the completion of cleavage furrow ingression (). Perhaps even more surprisingly, the D-box point mutant of cyclin B1, cyclin B1 R42A, was also rapidly degraded in the presence of the Aurora B inhibitor, while Histone 2B remained completely stable (; Fig. S2A and B). This suggests that Δ52–85-cyclin B1 and R42A-cyclin B1 abruptly turn into more efficient APC/CCdc20 substrates once the spindle checkpoint is inactivated in anaphase-arrested cells. These observation are potentially in line with a role for increasing Cdc20 activity in anaphase that we and others reported previously.Citation16 Alternatively, the APC/C activator Cdh1 becomes partially activated by Aurora B inhibition, even when a stabilized mutant of cyclin B1 maintains Cdk1 activity and represses APC/CCdh1. This explanation would imply that APC/CCdh1, when activated by Aurora B inhibition, would recognize a mutated D-box or a lysine mutant of cyclin B1 as its substrate although these mutants were not recognized as substrates by APC/CCdc20 before. Notably, cyclin B1 has no obvious KEN box, a typical APC/CCdh1 recognition motif.
Next, we investigated whether cyclin B1 mutant lacking the N-terminus, and thus a slightly larger region around the D-box APC/C recognition motif (ΔN-cyclin B1), did remain stable in mitosis even after override of the spindle checkpoint. Therefore, mitotic cells expressing R42A-cyclin B1, Δ52–85-cyclin B1 or ΔN-cyclin B1 were collected by mitotic shake-off. Next, these mitotically arrested cells were treated with the Mps1 inhibitor Reversine. Loss of mitotic Cdk1 phosphorylation of APC3, visible as a gel-shift, was used as a read-out for mitotic exit (). After Reversine treatment, cells expressing R42A-cyclin B1, or Δ52–85-cyclin B1, as implied by or data above, indeed dephosphorylated APC3 showing that they exited mitosis, and started to degrade the previously stabile cyclin B1 mutants (; compare lanes 3 and 4, with 7 and 8, respectively). Interestingly however, cells expressing ΔN-cyclin B1 remained firmly arrested in mitosis even after treatment with Mps1 inhibitor. This confirms that upon spindle checkpoint inactivation in anaphase, the APC/C becomes more active or starts to recognize a wider range of substrates, including R42A-cyclin B1 and Δ52–85-cyclin B1. The N-terminally deleted cyclin B1 mutant, which lacks a larger region around the D-box, is not seen as an APC/C substrate even after Mps1 inhibition in anaphase. This widening of substrate recognition by the APC/C after inhibition of Aurora B or Mps1 seems specific for late anaphase, because the cyclin B1 mutants arrested cells at a time point after they first inactivated the spindle checkpoint (see ; and refs. Citation8 and Citation18). This suggests there is an increase in catalytic APC/C activity in anaphase, providing that the spindle-checkpoint re-activation is overcome. Alternatively, in anaphase the APC/C, still bound to its activator Cdc20, gains a broadened substrate recognition as a consequence of Aurora B or Mps1 inhibition. A controlling function of Mps1 in spindle checkpoint re-activation is reminiscent of the situation in budding yeast.Citation22
Altogether our findings show that rapid and efficient cyclin B1 destruction is critical to prevent the spindle checkpoint from returning as a consequence of sister chromatid separation. Removal of cyclin B1 right after anaphase onset, at its latest, appears to be key to the coordination of sister chromatid separation with cell division. The efficiency by which cyclin B1 is removed may thus be crucial for genomic stability. A remarkable finding reported here is that either the catalytic activity or the substrate specificity of the APC/C seems to increase after Aurora B or Mps1 are inactivated in anaphase-arrested cells. As a result, poorly degradable cyclin B1 mutants become much better APC/C substrates. These findings suggest that, when cyclin B1 is not yet degraded and the APC/C activator Cdh1 should still be repressed, APC/CCdc20 can be activated by two processes: inactivation of the spindle checkpoint and progression into anaphase.Citation16 Such a mechanism, which would need to be investigated further, could for instance help to prevent spindle checkpoint re-activation in accidental cases when residual cyclin B1 remains during anaphase.
Interesting in this respect is that tumor cell lines co-depleted of Ube2S and UbcH10, two E2 enzymes subsequently required for nucleation and elongation of polyubiquitin-chains on APC/C substrates, may arrest similarly in a post-metaphase state.Citation20,Citation21,Citation23 Given the potential detrimental effects of the anaphase problem, in case it would occur, it will be interesting to investigate whether elevated cyclin B1 expression levels, or aberrancies in other factors that may influence the re-activation of the spindle checkpoint, could play a role in the occurrence of aneuploidy by causing the anaphase problem.
Materials and Methods
Cell culture and synchronization
The human osteosarcoma cell line U2OS, ecotropic-HeLa cells (ecotropic receptor on neomycin selection) and ecotropic-Phoenix cells were cultured in DMEM containing 8% FCS and antibiotics. Cells were plated on 35-mm glass bottom dishes (Willco Wells) or 9-cm Falcon dishes 24–48 h prior to synchronization. Cells were synchronized in mitosis by a 24 h thymidine block (Sigma, 2.5 mM final concentration) following release in the presence of nocodazole (Sigma, 830 nM final concentration) or taxol (Sigma, 1 μM final concentration) for 12–16 h. To obtain mitotic cells after expression of stable cyclin B1 mutants, no drug was added. Mitotic cells were collected by mitotic shake-off. G2-phase cells were collected 8 h after release from a thymidine block, after washing away any early mitotic cells. Drugs used as indicated: Mps1 inhibitor Reversine (#10004412, Cayman Chemicals, 75 nM final concentrat1ion); Proteasome inhibitor MG-132 (#13697, Cayman Chemicals, 5 μM final concentration); Aurora B inhibitor AZD-1152 (1 μM final concentration).
Plasmids and siRNA
The following Clontech expression plasmids have all been described and have been cloned using restriction sites as indicated: cyclin B1-Cherry (HindIII-BamHI), R42A-cyclin B1-Cerulean, ΔN-cyclin B1-Cerulean, INCENP-GFP and Geminin-Cherry (BglII-HindIII). Δ52–85-cyclin B1-Cerulean was constructed by PCR amplification of the coding sequence of the first 51 amino acids of cyclin B1 and cloning into cyclin B1-Cerulean using restriction sites XhoI (in the MCS) and Asp718 (located in the cyclin B1 cds). Viral plasmids pLIB-cyclin B1-Cherry, pLIB-Δ52–85-cyclin B1-Cerulean, ΔN-cyclin B1-Cerulean and pLIB-R42A-cyclin B1-Cerulean were constructed by cloning cyclin B1-Cerulean from Cerulean-N1 into pLIB-N1 using BglII-NotI restriction sites. All plasmids were sequence verified. H2B-ECFP was described beforeCitation8,Citation14,Citation24
Transfection
Cells were transfected with expression plasmids using a standard calcium phosphate transfection protocol or lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. For time-lapse microscopy, cells plated on Willco Well dishes were transfected with expression plasmids using calcium phosphate transfection as indicated in the following amounts: 0,29 µg R42A-cyclin B1-Cerulean, 0,29 µg Δ52–85-cyclin B1-Cerulean, 0,29 µg ΔN-cyclin B1-Cerulean. For transfection with lipofectamine 2000, 5 times less DNA was transfected. U2OS cells were co-transfected on 9 cm Falcon culture dishes, using 2,9 µg R42A-cyclin B1-Cerulean, 2,9 µg ΔN-cyclin B1-Cerulean or 2,9 µg Δ52–85-cyclin B1-Cerulean together with 1 µg of pBABE-Puro and selected with puromycin for 24 h (in the presence of thymidine as indicated).
Ecotropic Phoenix cells were transfected with 20 µg of pLIB-Δ52–85-cyclin B1-Cerulean or R42A-cyclin B1-Cerulean using standard calcium phosphate transfection. Viral supernatant was collected twice, 40 and 48 h after transfection. The supernatant was cleared through a 0.45 μm filter (Millipore). U2OS cells expressing the ecotropic receptor (from Johan Kuiken, NKI) were infected twice with the virus.
Western blots and immunoprecipitation
Cells were lysed in ELB+ (150 mM NaCl, 50 mM Hepes [pH 7.5], 5 mM EDTA, 0,3% NP-40, 10 mM β-glycerophosphate, 6% glycerol, 5 mM NaF, 1 mM Na2VO3, and Roche protease inhibitor cocktail) for 30 min. Lysates were cleared by centrifugation (13 000× g, 8 min, 4C). Protein levels were measured and equalized using standard Bradford assay (Biorad). For immunoprecipitations, lysates were incubated with 1–2 µg anti-APC4 antibody and 20 µg protein G Sepharose beads (Amersham Biosciences) for 4–8 h at 4C, while tumbling. After incubation, beads were 4 times washed with ice-cold ELB+ and dissolved in 50 µL sample buffer. Protein was separated on SDS-PAGE and blotted on nitrocellulose membranes. For membrane blocking and antibody incubation 4% ELK was used. Quantification of western blots was done with Image Lab software (Bio-Rad Laboratories).
Chromosome spreads
HeLa cells transfected with R42A-cyclin B1-Venus were 46 h later treated with 830 nM nocodazole for 30 min. Adherent cells and floating cells were harvested, centrifuged for 5 min at 1500 rpm, and supernatant was discarded. Subsequently, cells were resuspended in pre-warmed (at 37 °) 0.075 M KCl, while shaking constantly. Cells were incubated at 37 degrees for 10 min, a small volume of methanol/acetic acid (in a ratio of 3:1) was added dropwise, and cells were centrifuged for 5 min at 1500 rpm. The supernatant was discarded and the cell pellet was resuspended in a leftover of the supernatant. For fixation, 1 mL of the methanol/acetic acid solution was added while shaking and this suspension was incubated for 20 min at room temperature. Thereafter, the cells were centrifuged for 5 min at 1500 rpm. This fixation procedure was repeated 2 times. Lastly, the cell pellet was resuspended in methanol/acetic acid and the suspension was dropped from +/− 30 cm height onto clean microscope slides. The slides were air-dried, covered with DABCO/DAPI solution to stain the DNA, and protected by a glass coverslip.
Antibodies
For detection of proteins the following antibodies were used at the indicated dilution: mouse anti-CDC27 (#610455, 1:1000; BD Transduction), rabbit anti-geminin (FL-209, 1:1000; Santa Cruz), mouse anti-Cdc20 (E-7, SC-13162, 1:1000; Santa Cruz), goat anti-APC4 (C-18, sc-21414, 1:1000; Santa Cruz), mouse anti-cyclin B1 (GNS1, sc-245, 1:1000; Santa Cruz), mouse anti-Mad2 (K0167–3, 1:1000; MBL), goat anti-Actin (I-19, sc-1616, 1:1000; Santa Cruz), rabbit anti-APC10 (611501/2, 1:1000; BioLegend) and mouse anti-BubR1 (MAB3612, 1:1000; Chemicon), rabbit anti-Aurora A/ AIK (#3092, 1:1000; Cell Signaling Technology). Secondary PO-conjugated antibodies were obtained from DAKO.
Time-lapse fluorescence microscopy
U2OS cells transfected with indicated plasmids were followed by fluorescence time-lapse microscopy.Citation8,Citation14,Citation24 Acquisition of DIC and fluorescence images started 46 h after transfection on a microscope (Zeiss Axio Observer Z1) in a heated culture chamber (5% CO2 at 37 °C) using DMEM with 8% FCS and antibiotics. The microscope is equipped with a LD 0.55 condenser and 40× NA 1.40 Plan-Apochromat Oil DIC objective and CFP/ YFP and GFP/ HcRed filter blocks (Zeiss) to select specific fluorescence. Images were taken using Zeiss software (AxioVision Rel. 4.8.1) with a charge-coupled device camera (Hamamatsu ORCA R2 Black and White CCD) at 100-ms exposure times. For quantitative analysis of degradation, MetaMorph software (Universal Imaging) and Excel (Microsoft) were used. Captured images were processed using PhotoShop and Illustrator software.
| Abbreviations: | ||
| APC/C | = | Anaphase-Promoting Complex or Cyclosome |
| DIC | = | differential interference contrast |
| NEB | = | nuclear envelope breakdown |
Additional material
Download Zip (1.2 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by project grants from NWO ALW (Vidi, R.W.); KWF project grants NKI-2007-3789 (L.C. and J.O.) and NKI-2008-4135 (E.V. and B.t.R.), an EMBO fellowship (C.R.), and a Human Frontiers Science Program grant RGP0053/2010 (M.B., R.W.).
References
- Wheatley SP, Hinchcliffe EH, Glotzer M, Hyman AA, Sluder G, Wang Yl. CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J Cell Biol 1997; 138:385 - 93; http://dx.doi.org/10.1083/jcb.138.2.385; PMID: 9230080
- Hagting A, Den Elzen N, Vodermaier HC, Waizenegger IC, Peters JM, Pines J. Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J Cell Biol 2002; 157:1125 - 37; http://dx.doi.org/10.1083/jcb.200111001; PMID: 12070128
- Stemmann O, Zou H, Gerber SA, Gygi SP, Kirschner MW. Dual inhibition of sister chromatid separation at metaphase. Cell 2001; 107:715 - 26; http://dx.doi.org/10.1016/S0092-8674(01)00603-1; PMID: 11747808
- Holland AJ, Taylor SS. Cyclin-B1-mediated inhibition of excess separase is required for timely chromosome disjunction. J Cell Sci 2006; 119:3325 - 36; http://dx.doi.org/10.1242/jcs.03083; PMID: 16868023
- Shindo N, Kumada K, Hirota T. Separase sensor reveals dual roles for separase coordinating cohesin cleavage and cdk1 inhibition. Dev Cell 2012; 23:112 - 23; http://dx.doi.org/10.1016/j.devcel.2012.06.015; PMID: 22814604
- Yamano H, Gannon J, Hunt T. The role of proteolysis in cell cycle progression in Schizosaccharomyces pombe. EMBO J 1996; 15:5268 - 79; PMID: 8895572
- Wolf F, Wandke C, Isenberg N, Geley S. Dose-dependent effects of stable cyclin B1 on progression through mitosis in human cells. EMBO J 2006; 25:2802 - 13; http://dx.doi.org/10.1038/sj.emboj.7601163; PMID: 16724106
- Clijsters L, Ogink J, Wolthuis R. The spindle checkpoint, APC/C(Cdc20), and APC/C(Cdh1) play distinct roles in connecting mitosis to S phase. J Cell Biol 2013; 201:1013 - 26; http://dx.doi.org/10.1083/jcb.201211019; PMID: 23775192
- Vázquez-Novelle MD, Mirchenko L, Uhlmann F, Petronczki M. The ‘anaphase problem’: how to disable the mitotic checkpoint when sisters split. Biochem Soc Trans 2010; 38:1660 - 6; http://dx.doi.org/10.1042/BST0381660; PMID: 21118144
- Vázquez-Novelle MD, Petronczki M. Relocation of the chromosomal passenger complex prevents mitotic checkpoint engagement at anaphase. Curr Biol 2010; 20:1402 - 7; http://dx.doi.org/10.1016/j.cub.2010.06.036; PMID: 20619651
- Rattani A, Vinod PK, Godwin J, Tachibana-Konwalski K, Wolna M, Malumbres M, Novák B, Nasmyth K. Dependency of the spindle assembly checkpoint on Cdk1 renders the anaphase transition irreversible. Curr Biol 2014; 24:630 - 7; http://dx.doi.org/10.1016/j.cub.2014.01.033; PMID: 24583015
- Vázquez-Novelle MD, Sansregret L, Dick AE, Smith CA, McAinsh AD, Gerlich DW, Petronczki M. Cdk1 inactivation terminates mitotic checkpoint surveillance and stabilizes kinetochore attachments in anaphase. Curr Biol 2014; 24:638 - 45; http://dx.doi.org/10.1016/j.cub.2014.01.034; PMID: 24583019
- Kamenz J, Hauf S. Slow checkpoint activation kinetics as a safety device in anaphase. Curr Biol 2014; 24:646 - 51; http://dx.doi.org/10.1016/j.cub.2014.02.005; PMID: 24583014
- Mirchenko L, Uhlmann F. Sli15(INCENP) dephosphorylation prevents mitotic checkpoint reengagement due to loss of tension at anaphase onset. Curr Biol 2010; 20:1396 - 401; http://dx.doi.org/10.1016/j.cub.2010.06.023; PMID: 20619650
- Kitagawa M, Fung SY, Hameed UF, Goto H, Inagaki M, Lee SH. Cdk1 coordinates timely activation of MKlp2 kinesin with relocation of the chromosome passenger complex for cytokinesis. Cell Rep 2014; 7:166 - 79; http://dx.doi.org/10.1016/j.celrep.2014.02.034; PMID: 24656812
- Gurden MD, Holland AJ, van Zon W, Tighe A, Vergnolle MA, Andres DA, Spielmann HP, Malumbres M, Wolthuis RMF, Cleveland DW, et al. Cdc20 is required for the post-anaphase, KEN-dependent degradation of centromere protein F. J Cell Sci 2010; 123:321 - 30; http://dx.doi.org/10.1242/jcs.062075; PMID: 20053638
- van Zon W, Ogink J, ter Riet B, Medema RH, te Riele H, Wolthuis RMF. The APC/C recruits cyclin B1-Cdk1-Cks in prometaphase before D box recognition to control mitotic exit. J Cell Biol 2010; 190:587 - 602; http://dx.doi.org/10.1083/jcb.200912084; PMID: 20733055
- D’Angiolella V, Mari C, Nocera D, Rametti L, Grieco D. The spindle checkpoint requires cyclin-dependent kinase activity. Genes Dev 2003; 17:2520 - 5; http://dx.doi.org/10.1101/gad.267603; PMID: 14561775
- Yang Q, Ferrell JE Jr.. The Cdk1-APC/C cell cycle oscillator circuit functions as a time-delayed, ultrasensitive switch. Nat Cell Biol 2013; 15:519 - 25; http://dx.doi.org/10.1038/ncb2737; PMID: 23624406
- Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, Rape M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci U S A 2009; 106:18213 - 8; http://dx.doi.org/10.1073/pnas.0907887106; PMID: 19822757
- Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, Pines J, Venkitaraman AR. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol 2009; 11:1363 - 9; http://dx.doi.org/10.1038/ncb1983; PMID: 19820702
- Palframan WJ, Meehl JB, Jaspersen SL, Winey M, Murray AW. Anaphase inactivation of the spindle checkpoint. Science 2006; 313:680 - 4; http://dx.doi.org/10.1126/science.1127205; PMID: 16825537
- Van Zon W, Wolthuis RMF. unpublished observations.
- Voets E, Wolthuis RMF. MASTL is the human orthologue of Greatwall kinase that facilitates mitotic entry, anaphase and cytokinesis. Cell Cycle 2010; 9:3591 - 601; http://dx.doi.org/10.4161/cc.9.17.12832; PMID: 20818157