Abstract
The role of JAK signaling in cell cycle transit and maintenance of genomic stability was determined in HL-60 human myeloblastic leukemia cells. We have previously reported that a pan-JAK inhibitor caused ERK-dependent endoreduplication. In the current study we find that JAK inhibition caused nuclear re-localization of RAF-1, which could be inhibited by RAF inhibitor GW5074. GW5074 also inhibited JAK inhibitor-induced appearance of nuclear phosphorylated RAF-1(pS621RAF) and MEK, and it inhibited the JAK inhibitor-induced co-immunoprecipitation of nuclear RAF-1 and MEK. JAK inhibition also increased nuclear BubR1 phosphorylation, which was diminished by RAF inhibitor GW5074. RAF-1 and BubR1 in the nucleus co-immunoprecipitated; and GW5074 eliminated this. Furthermore, inhibiting RAF with GW5074 blocked the pan-JAK inhibitor-induced endoreduplication. These data thus show that JAK inhibition causes nuclear re-localization and phosphorylation of RAF and MEK where RAF binds BubR1 with ensuing nuclear RAF-dependent BubR1 phosphorylation. Inhibiting RAF inhibited this and endoreduplication. The results suggest that there is a JAK/RAF/MEK/BubR1 axis that can regulate genomic stability. In this hypothetical model JAK suppresses RAF/MEK phosphorylation and nuclear re-localization, but JAK inhibition induces the phosphorylations and re-localization with association of RAF and phosphorylated BubR1 in the nucleus leading to endoreduplication.
Introduction
In many cancers disruption of cell cycle transit and genomic instability are involved in cell transformation. For many tumors including leukemias cytokine regulated signaling through JAKs is aberrant. The function of such signaling is thus of significant interest in the pathology of cells whose growth and differentiation are governed by cytokines.
Janus family tyrosine kinases (JAKs) are associated with cytokine and growth factor receptors.Citation1 Activation of these receptors via ligand binding induces activation of the receptor associated JAKs through cross-phosphorylation. The tyrosine phosphorylated sites become docking sites for src homology 2 (SH2) domains of signaling proteins. Prominent among these are the STATs. Receptor recruited STATs are phosphorylated by JAKs, homo- or heterodimerize and rapidly translocate to the nucleus to induce target gene transcription.Citation2 The JAK signaling pathways mediate both anti-proliferative response following interferon stimulation, which is predominantly mediated by STAT1 and cellular proliferation in response to cytokines and growth factors, which is mediated by STAT3 and 5.
We have recently reported that a pan-JAK inhibitor caused endoreduplication in HL-60 cells. The JAK inhibitor also increased ERK phosphorylation; and using MEK inhibitor PD98059 prevented endoreduplication in JAK inhibitor treated cells.Citation3 The JAK/STAT pathways have been shown to negatively cross-talk with MAPK signaling. ERK and STAT1 have been reported to co-immunoprecipitate.Citation4 The antagonistic behavior of STAT1 and ERK was demonstrated with IFNα inducing STAT1 phosphorylation and at the same time decreasing ERK and MEK activation. Vice versa, RAS/ERK signaling could protect tumor cells from apoptosis by INFαCitation5 also indicating a negative crosstalk between ERK and STAT1. MAPKs have also been reported to interact with STAT3 and STAT5.Citation6–Citation8 A negative cross talk between c-RAF and the JAK/STAT pathway has been reported by Loucks et al.Citation9 Inhibiting MEK triggered STAT-1 expression and inhibiting JAKs using the same inhibitor as we used in the current study restored c-RAF mediated survival in neurons.
The prototypic mitogenic MAPK signaling through a RAF/MEK/ERK cascade is initiated by activated growth factor receptors, at the cell membrane.Citation10 RAF is activated by phosphorylation. S621 is an activating phosphorylation site.Citation11 Subsequently, activated RAF phosphorylates and activates MEKs that in turn phosphorylate ERKs. ERKs then phosphorylate a number of cytoplasmic and nuclear targets, resulting in changes in transcription and translation.Citation12 While these prototypic RAF activations were associated with G0 to G1 transition, RAF was also shown to be hyperphosphorylated during mitosis.Citation13 Specifically, RAF has been found to physically interact with retinoblastoma protein (RB) in the nucleus and thereby inhibiting RB's suppression of cell cycle progression.Citation14 In addition, RAF and RAF kinase inhibitory protein (RKIP) have been shown to regulate the spindle checkpoint via Aurora B during G2/M transition.Citation15 This mitotic checkpoint is known to be regulated by BubR1.Citation16 BubR1 is a kinase binding at kinetochoresCitation17 that regulates the Anaphase Promoting Complex (APC) that controls mitosis.Citation18 It is a phosphoprotein that is transcriptionally regulated by p53.Citation19 It has been found to have various cooperative partners, including PCAF,Citation17 polo-like kinase 1,Citation20 and aurora B.Citation21 Interestingly its deficiency disrupts megakaryopoiesis, a process where ploidy increases to generate megakaryocytes.Citation22 It is thus a likely candidate for controlling genomic stability.
Because our previously observed JAK inhibitor-induced endoreduplication appeared to be ERK dependent, and because of a suggested role for RAF during mitosis and possible nuclear localization of RAF, we hypothesized RAF would migrate into the nucleus and potentially regulate a mitotic checkpoint during JAK inhibitor-induced endoreduplication.
Results
Inhibition of JAKs induces RAF-/pS621-RAF-1 nuclear translocation.
To investigate whether RAF translocates into the nucleus during JAK inhibitor-induced endoreduplication we probed for RAF and pS621-RAF in western analysis of nuclear fractions from cells treated with JAK inhibitor for 48 and 72 hours. JAK inhibition induced RAF nuclear re-localization after 48 and 72 hours which could be inhibited by RAF inhibitor GW5074 ( shows the western of the nuclear fraction). As expected, shRNA targeting RAF also eliminated the nuclear signal. Blots were probed for lamin A as a lane loading control. The nuclear translocation of RAF resulted in a decrease of RAF in the cytosol when compared to untreated HL-60 cells ( shows westerns of the nuclear and cytosolic fractions). Similarly, we detected phospho S621-RAF appearing in the nucleus following 48 and 72 hours of treatment with the JAK inhibitor ( shows westerns detecting signal in the nuclear but not cytosolic fractions). The JAK inhibition-induced appearance of nuclear S621 phosphorylated RAF was inhibited by GW5074. The JAK inhibitor did not change RAF phosphorylation in the cytosol. Lamin A and HSP were probed to demonstrate equal loading of nuclear and cytosolic fractions, respectively. Inhibition of JAKs thus caused RAF phosphorylation at S621 and translocation from the cytosol to the nucleus.
Inhibition of JAKs induces MEK nuclear translocation.
The RAF nuclear localization motivated interest in determining whether the downstream MEK could also be found in the nucleus upon JAK inhibition. 48 and 72 hours post JAK inhibitor treatment we detected phosphorylated MEK in the nucleus which could be inhibited by RAF inhibitor GW5074 (). To determine whether MEK and RAF-1 physically interact in the nucleus we immunoprecipitated MEK and probed for RAF-1 in a western analysis. shows that the JAK inhibitor induced a GW50745 sensitive MEK and RAF-1 interaction in the nucleus following 48 and 72 hours of treatment. JAK inhibition thus caused pMEK nuclear re-localization which is dependent on RAF activation and the MEK and RAF in the nucleus co-immunoprecipitate.
Inhibition of JAKs induces BubR1 phosphorylation which is RAF dependent.
To investigate whether JAK inhibitor-induced endoreduplication affects G2/M cell cycle check point proteins, we determined BubR1 phosphorylation. 48 and 72 hours post JAK inhibitor treatment, BubR1 was phosphorylated in nuclear fractions ( shows a western of BubR1 and gel mobility retardation of the phosphorylated BubR1). GW5074 treatment inhibited this BubR1 phosphorylation in response to JAK inhibition. JAK inhibition thus caused phosphorylation of the BubR1 mitotic checkpoint regulator dependent on nuclear activated RAF.
Inhibition of JAKs causes nuclear RAF and BubR1 association.
To determine if RAF complexed with BubR1 in the nucleus, nuclear BubR1 was immunoprecipitated and subjected to western analysis probing for RAF. Cells were treated with JAK inhibitor or JAK inhibitor plus GW5074 for 48 or 72 hrs. Nuclei were isolated and analyzed ( shows the western probed for RAF). RAF co-immunoprecipitated with BubR1 in JAK inhibitor treated cells but not JAK inhibitor plus GW5074 treated cells. JAK inhibition thus caused nuclear RAF and BubR1 co-immunoprecipitation dependent on RAF activation, which was shown above to equate to its nuclear translocation with JAK inhibition.
To visualize and corroborate nuclear RAF and BubR1 association, immunofluorescence microscopy of cells treated with JAK inhibitor for 48 and 72 hrs versus untreated was performed. Cells were immunofluorescently stained for RAF (green), BubR1 (red), nuclear DNA (blue). As expected in untreated cells, the RAF signal is relatively bright in the cytoplasm and dark in the nucleus. The RAF images show its JAK inhibitor-induced movement into the nucleus by 72 hrs and the merged RAF and BubR1 images confirm their nuclear co-localization ().
If JAK inhibition affected the BubR1 mitotic checkpoint regulator and ultimately activated the mitotic exit checkpoint to cause tetraploidy by failure of cytokinesis, then one might expect that cyclin B1 would be stabilized when the checkpoint is activated. To investigate this, cyclin B1 expression in cells treated with JAK inhibitor (for 72 hrs.) was measured by western analysis (). Expression in JAK inhibitor-treated cells was enhanced, consistent with anticipation. In contrast cells treated with JAK inhibitor plus GW5074 did not show this enhancement. The results are consistent with JAK inhibitor-induced activation of the mitotic exit checkpoint and stabilization of cyclin B1.
If cyclin B1 were stabilized as suggested above, then one might anticipate that release from mitotic arrest of JAK inhibitor treated cells would be slower than for untreated cells. In order to exit M, B1 must be degraded and a higher B1 expression might thus be expected to result in slower exit. Checkpoint stabilization by BubR1 expression, for example, has been found to do this.Citation15 To test this, proliferating cells were treated with nocodazole or nocodazole plus JAK inhibitor to cause mitotic arrest and then released from nocodazole. JAK inhibitor treated cells continued to be treated with JAK inhibitor. The percentage of cells in G2/M was measured by flow cytometry during the nocodazole block and thereafter (). Both JAK inhibitor untreated and treated cells showed a similar rate of accumulation in G2/M, demonstrating that the JAK inhibitor had no discernable effect on cell cycle rates. After release from nocodazole, the cells treated with JAK inhibitor had a slower exit from G2/M. JAK inhibition thus affected the BubR1 mitotic checkpoint regulator in a RAF dependent manor with anticipated effects on cyclin B1 and the mitotic exit checkpoint.
Inhibiting RAF with GW5074 blocks JAK inhibitor-induced endoreduplication.
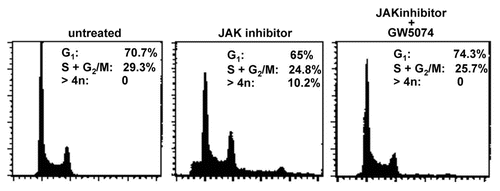
If JAK inhibitor induced RAF activation and nuclear re-localization, nuclear RAF association with BubR1, and its phosphorylation were a causal sequence of events for endoreduplication; then inhibition of this sequence by GW5074 would also be expected to inhibit JAK inhibitor-induced endo reduplication as well. To test this, cells were treated with JAK inhibitor or JAK inhibitor plus GW5074 for 48 hrs. DNA histograms of the resulting cells were generated by flow cytometry. RAF inhibition almost completely blocked the JAK inhibitor-induced endoreduplication ( shows DNA histograms). Cell populations treated with JAK inhibitor had obvious cells with greater than 4n DNA content and an evident 8n DNA histogram peak, but the cell population treated with JAK inhibitor plus GW5074 had no discernable cells with greater than 4n DNA. Of relevance, the DNA histogram of cells treated with the combination of JAK inhibitor plus the GW5074 RAF inhibitor showed no G1 arrest, nor —as would be anticipated—did cells treated with just a single agent (R. Tassef and A. Yen, not shown single and shown combined treatment histograms are indistinguishable); hence of course the lack of endoreduplication with GW5074 was not attributable to a simple G1 cell cycle block. RAF inhibition thus also inhibited JAK inhibitor-induced endoreduplication.
Discussion
In summary, we find that inhibition of JAKs leads to nuclear localization and phosphorylation of RAF-1 and MEK-1 and RAF-dependent BubR1 phosphorylation and endoreduplication. In addition, we demonstrate that RAF-1 co-immunoprecipitates with MEK-1 and BubR1 in the nucleus due to JAK inhibition. Inhibiting RAF with GW5074 inhibited the RAF nuclear re-localization, S621 phosphorylation and association with MEK and BubR1. GW5074 also inhibited endoreduplication, consistent with dependence of the induced endoreduplication on these RAF events. The data are potentially consistent with a model in which JAKs suppress RAF nuclear re-localization and phosphorylation and JAK inhibition allows RAF nuclear re-localization and phosphorylation; the nuclear RAF binds to BubR1 which becomes phosphorylated and affects the APC/mitotic checkpoint to result in endoreduplication.
We provide novel evidence for nuclear localization of RAF and MEK during endoreduplication. Although the historical perception of RAF is as a cytosolic signaling molecule, RAF has been found in the nucleus before. For example, RAF has been found to physically interact with RB in the nucleus.Citation13 In addition, RAF and RAF kinase inhibitory protein (RKIP) have been shown to regulate the spindle checkpoint via Aurora B during G2/M transition.Citation14 Tyrosine phosphorylated ERK was also found in proximity to mitotic spindles when relocating from the nucleus to the Golgi complex during G2 and mitosis.Citation23 RAF is also driven into the nucleus by retinoic acid when it induces cell differentiation.Citation24
BubR1 phosphorylation appears to be associated with endoreduplication in the present studies. We have previously reported that inhibiting JAKs causes enhanced ERK phosphorylation and endoreduplication which could be prevented by the MEK inhibitor PD98059.Citation3 Endoreduplicating cells underwent mitosis as determined by histone 3 phosphorylation, an event occurring early during mitosis. However, the cells failed to divide. Here, we report that JAK inhibitor resulted in BubR1 phosphorylation. BubR1 is a cell cycle M phase check point protein and is involved in inhibiting the anaphase promoting complex (APC). In addition, the BubR1 phosphorylation was inhibited by RAF inhibitor GW5074. BubR1, activated ERK and MEK have been found to physically interact with each other and localize to spindle poles during mitosis.Citation25 BubR1 knock down and BubR1 deficiency both resulted in enhanced MEK and ERK activation during mitosis. While this suggests a negative regulatory relationship, we found that after GW5074, MEK inhibition was associated with inhibited BubR1 phosphorylation. It is therefore also possible, that BubR1 was phosphorylated in response to the genomic instability induced by the JAK inhibitor rather than in response to nuclear RAF and MEK. However, the genomic instability, namely endoreduplication, induced by the JAK inhibitor appears to be caused by RAF activation since the JAK inhibitor-induced endoreduplication could be blocked by RAF inhibitor GW5074. Previously we observed that MEK inhibitor PD98059 could inhibit signs of JAK inhibitor-induced endoreduplication. The effect of PD98059 in reducing multi-nucleation and spindle disturbance was also observed by others when using fumarylacetoacetate to induce endoreduplication.Citation26 In our present study, the data suggest that RAF and MEK activation act together as upstream events for endoreduplication.
While MEK, RAF and ERK are important in cell proliferation, if uncontrolled by proteins such as BubR1, RB or MAD2, these growth signals can potentially result in genomic instabilities such as endoreduplication. RAF-1 has been found to physically bind to RB upon serum stimulation of quiescent fibroblasts and inactivate RB's suppressive function on E2F1 transcription and cell proliferation.Citation13 The JAK inhibition with subsequent RAF and MEK nuclear localization might hence inactivate RB in HL-60 cells. Because HL-60 cells are p53 negative, which makes cells vulnerable to genomic instability,Citation27 an inactivation of RB could be, at least in part, causal to the endoreduplication observed upon JAK inhibition.
In sum, we provide novel evidence for RAF and MEK activation and nuclear localization during JAK inhibitor-induced endoreduplication. Moreover, we found that nuclear RAF is physically associated with the phosphorylated BubR1 APC/mitotic checkpoint regulator. The precise mechanism on how the JAK/STAT pathway interferes with the MAPK pathway is not clear. However, because activated STATs have been found to bind to ERK one could speculate there is a sequestering role for activated STATs. During JAK/STAT inhibition and hence lack of MAPK sequestering in the cytosol, MAPKs appear to migrate into the nucleus.
Materials and Methods
Cell culture.
HL-60 human myeloblastic leukemia cells were grown in RPMI 1640 supplemented with 5% heat-inactivated fetal bovine serum (both: Invitrogen, Carlsbad, CA) and 1x antibiotic/antimicotic (Sigma, St. Louis, MO) in a 5% CO2 humidified atmosphere at 37°C. JAK inhibitor I (targeting JAK1, JAK2, JAK3 and Tyk2) (Calbiochem, La Jolla, CA) were administered from stock solutions in DMSO with a final concentration of 2 mM, 10 µM. RAF inhibitor GW5074 (Sigma, St. Louis, MO) was administered from a stock solution in DMSO 1 h prior to treatment with JAK inhibitors. Experimental cultures were initiated at a density of 0.2 × 106 cells/ml and assayed 24, 48 and 72 h post treatment. Viability was monitored by 0.2% trypan blue (Invitrogen, Carlsbad, CA) exclusion and routinely exceeded 95% prior to drug administration. The GW5074 did not cause a G1 cell cycle block in these hematopoietic cells.
For nocodazole treatment experiments, flow cytometry was used to measure cells with G2/M DNA content. Parallel cultures of cells were co-treated with nocodazole (100 ng/ml) and JAK inhibitor or just nocodazole, and their DNA histogram was measured at different times subsequently. The percentage of cells with 4n DNA at times 0, 12, 24 h showed the pattern of accumulation of cells in G2/M. (The majority of untreated or JAKI- treated cells arrested in G2/M phases after being treated with nocodazole for 24 h). After 24 h of nocodazole treatment, cells were resuspended in fresh medium with or without JAK inhibitor alone in the cultures for another 12 h and then harvested for analysis of the DNA histogram by flow cytometry (36 h of culture).
Western blotting.
Protein was extracted from cells using a 1% SDS lysis buffer. DNA was removed by centrifugation at 13,000 rpm at 4°C for 10 min. Protein concentration was determined by measuring the absorbance at 585 nm of proteins in a Bradford assay (Sigma, St. Louis, MO). 15 µg of protein was loaded on a 12% tris-HCL precast gel (Biorad, Hercules, CA). Following electrophoresis at 120 V for 2 h, protein was electro-transferred onto an Imobilon-P membrane (Millipore, Bedford, MA) for 2 h at 90 V. Membranes were blocked in 5% non-fat milk in TBS-tween and probed with anti- MAPK p44/42, β-actin (Cell Signaling, Danvers, MA) or STAT3 pY705 (BD Biosciences, San Jose, CA), respectively. To detect these probes by ECL, HRP-conjugated anti-rabbit and anti-mouse antibodies were used as secondary antibodies, respectively. Blots were incubated with Detection Reagents 1 and 2 (Amersham Biosciences, Buckinghamshire, United Kingdom) and visualized using blue sensitive X-ray film (LPS, Rochester, NY). Blots were stripped and re-probed for β-actin as a loading control. All blots were repeated at least 3 times.
Isolation of various cellular fractions.
The nuclear and cytosol fractions were isolated using the nuclear/cytosol fractionation kit from BioVision (BioVision, Mountain View, CA, USA), or by following procedure. In brief, cells (1 × 107), after different treatments, were incubated with 1% Triton X-114 lysis buffer (1% Triton X-114, 25 mM Tris, pH 7.5, 20 mM MgCl2, 150 mM sodium chloride, 1 mg/ml aprotinin and 1 mg/ml leupeptin) on ice for 30 min and then homogenized by passing through a 25-gauge needle for 45 passages. After centrifuging at 280 g for 15 min, supernantant was collected as the cytosol fraction. The precipitated nuclei were then lysed with nuclear lysis buffer (50 mM Tris-Cl, pH 7.6, 10 mM EDTA, 1% sodium dodecyl sulfate, 1 mM phenylmethylsulphonyl fluoride, 1 mg/ml aprotinin and 1 mg/ml leupeptin) on ice for 10 min. The nuclear extract was collected by re-centrifuging at 280 g for 15 min. The supernatants were collected and subjected to centrifugation again at 16,000x g for 30 min. Subsequently, the supernatants were collected as the cytosolic fraction.
Immunoprecipitation.
After different treatments, the nuclear fraction from each sample was isolated and the total protein concentration in each fraction was normalized. Subsequently, the nuclear fraction was immunoprecipitated with anti-MEK Ab overnight in a cold room. Immunoprecipitates were collected with protein G-sepharose and separated on a 10% SDS-PAGE gel. Raf or MEK was then detected by western blotting with anti-Raf Ab or anti-MEK Ab, respectively.
Immunofluorescence.
After treatments, cells seeded on a cover glass were fixed with 3.7% paraformaldehyde in 1x PBS for 10 min. Following permeabilization with 0.2% Triton X-100 for 5 min at room temperature, cells were incubated with anti-Raf1 or -BubR1 primary antibody and then incubated with a FITC-conjugated anti-rabbit secondary antibody (Invitrogen) or cyanine Cy5 conjugated anti-donkey secondary antibody (Jackson Immunpo Research) as well as DAPI (4′,6-diamidino-2-phenylindole). The cells were visualized with a Zeiss Axio Imager Z microscope. The images were captured using the AxioVision Rel. 4.6 software (Carl Zeiss MicroImaging).
DNA histograms.
After different treatments, 0.5 × 106 cells were centrifuged to a pellet at 1,000 rpm for 5 min. and permeablized with 90% methanol for 20 min. Samples were washed 2x in 1 ml PBS and stained with 200 ul PBS containing 5 ug/ml DAPI (from a 0.2 mg/ml solution in ddH2O, Polysciences, Warrington, PA). Cells were incubated for 1 h and analyzed by flow cytometry (BD LSRII, BD Biosciences, San Jose, CA). Doublets were identified by a DAPI signal width versus area plot and excluded from analysis.
Retroviral construct delivery.
Small hairpin RNA (shRNA) lentiviruses were generated following 293T transfection with the helper DNAs using Fugene HD (Roche). Forty-eight hours later, virus containing supernatants were filtered through 0.45 um syringe filters. Cells were infected prior to treatments.
Figures and Tables
Figure 1 Inhibiting JAKs causes nuclear localization of RAF-1, pS621-RAF and p-MeK. Western blots in (A) (western blot of nuclear fraction probed for RAF in JAK inhibitor (JAK) treated (48 and 72 h) and JAK inhibitor plus shRNA targeting RAF or GW5074 RAF inhibitor treated (72 h) cells) show nuclear localization of RAF-1 upon 48 and 72 hours of treatment with a pan-JAK inhibitor. The nuclear migration of RAF-1 was inhibited in the presence of shRNA targeting RAF-1 and RAF inhibitor GW5074. Blots were reprobed for Laminin A as a lane loading control. Occurrence of RAF-1 in the nucleus in response to JAK inhibition coincided with a decreased amount of RAF-1 protein in the cytosol as shown in (B) (western blots of cytosolic (upper) and nuclear (lower) fractions probed for RAF). (C) (western blot for nuclear and cytosolic fractions of cells treated with JAK inhibitor (48 and 72 h) or JAK inhibitor plus GW5074 RAF inhibitor (72 h)) shows pS621-RAF-1 nuclear localization in response to JAK inhibition for 72 hours which was blocked in the presence of RAF inhibitor GW5074. pS621-RAF-1 protein levels remained unchanged in the cytosol.
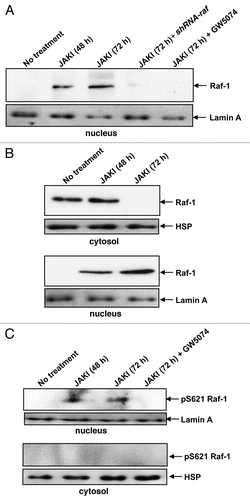
Figure 2 JAK inhibition caused nuclear localization of p-MEK-1 and nuclear interaction of MEK-1 and RAF-1. Following 48 or 72 hours of JAK inhibition we observed nuclear P-MEK-1. RAF inhibitor GW5074 blocked p-MEK-1 nuclear re-localization (A, western blot of nuclear fraction probed for p-MEK). Blots were reprobed with Laminin A to demonstrate equal protein loading. (B) shows physical interaction of RAF and MEK in the nucleus in response to JAK inhibitor treatment for 48 or 72 hours. The immunoprecipitation was performed using an anti-MEK-1 antibody followed by a western Blot probing for RAF-1. Blots were also probed for MEK-1 to control for uneven MEK-1 expression.
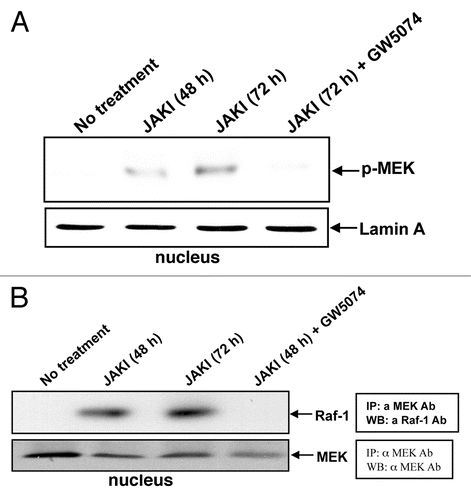
Figure 3 Inhibiting JAKs causes BubR1 phosphorylation. APC/mitotic check point protein BubR1 was phosphorylated upon JAK inhibitor treatment for 48 or 72 hours and the BubR1 phosphorylation was decreased by RAF inhibitor GW5074. We included Jurkat cells as a positive control. The western blot of the nuclear fraction shows BubR1 and phosphorylation-induced gel mobility retarded BubR1.
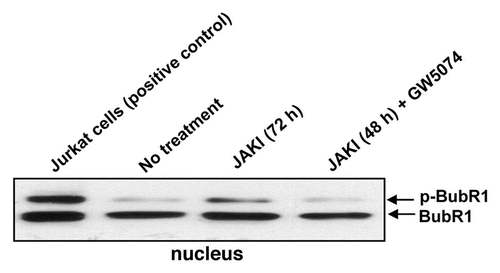
Figure 4 JAK inhibition causes nuclear BubR1 and RAF-1 association. Immunoprecipitation of BubR1 from nuclear extracts of cells treated with JAK inhibitor or JAK inhibitor plus GW5074 for 48 or 72 hours and western blotting for RAF-1 show that BubR1 and RAF co-immunoprecipitate and that GW5074 eliminates the association. (A) Western blot of BubR1 co-immunoprecipitating proteins probed for RAF in the nuclear fraction from cells treated with JAK inhibitor or JAK inhibitor plus GW5074 for 48 or 72 h. Immunofluorescent microscopy of untreated (top row), or JAK inhibitor treated (48 hours, middle row, 72 hours, bottom row) cells stained for RAF (left to right columns), BubR1, nuclear DNA and merged image. (B) Immunofluorescence images of the same field.
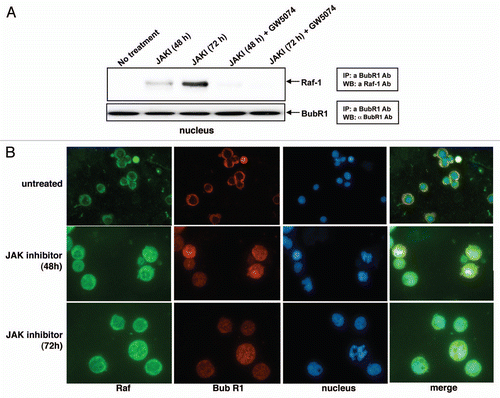
Figure 5 Statistical Analysis. three independent repeats were conducted in all experiements. Error bars represent these repeats. A Student's T test was used and a p value of <0.05 was considered significant. JAK inhibition affects mitotic checkpoint. JAK inhibitor enhanced cyclin B1 expression in the population, but JAK inhibitor plus GW5074 did not. Cells were treated with JAK inhibitor or JAK inhibitor plus GW5074 and subject to western analysis of cyclin B1. Actin was a lane loading control. (A). JAK inhibitor retarded mitotic exit. Cells were treated with nocadazole or nocadazole plus JAK inhibitor. The nocadazole induced a G2/M arrest measured by flow cytometry. Both JAK inhibitor untreated and treated populations arrested with indistinguishable rates. Both populations were then released from nocadazole, one in the absence of JAK inhibitor and the other continued to be JAK inhibitor treated. The JAK inhibitor cells showed a slower exit from mitosis, that is a higher mitotic index, 12 hours after release. The asterisk indicates statistical significance at p < 0.05, n = 3. (B, Left to right shows the percentage of cells with G2/M DNA content at 0, 12 and 24 hrs of treatment with nocodazole or nocodazole plus JAK inhibitor and at 12 hrs after release from the nocodazole).
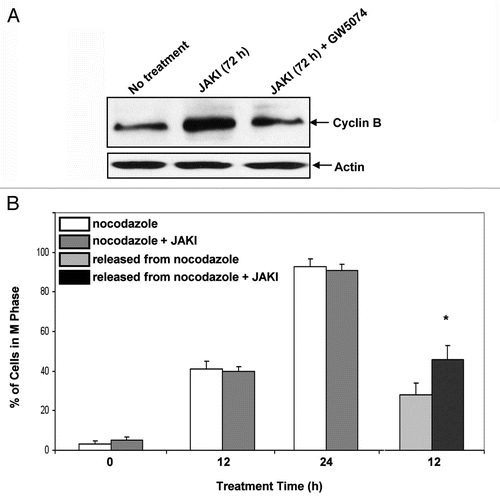
Figure 6 Inhibiting RAF blocks JAK inhibitor-induced endoreduplication. Treatment (48 hr) with JAK inhibitor caused endoreduplication as indicated by G2/M accumulation and cells with a DNA content of greater than 4n. RAF inhibitor GW5074 almost completely inhibited JAK inhibitor-induced endoreduplication restoring the DNA histogram to be comparable to the untreated control. Flow cytometric DNA histograms for untreated, JAK inhibitor treated and JAK inhibitor plus GW5074 treated cells (left to right parts) are shown.
Acknowledgements
Supported by grants from USPHS NIH (CA100498) (C-YC), NSF (CBET-0846876)(JDY), and NIH (CA33505) and NYSTEM NY Dept. Health (AY). We are grateful to Dr. Rodica P. Bunaciu for critical reading, editing and advice.
References
- Yamaoka K, Saharinen P, Pesu M, Holt VE 3rd, Silvennoinen O, O'Shea JJ. The Janus kinases (JAKs). Genome Biol 2004; 5:253
- Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003; 101:2940 - 2944
- Reiterer G, Yen A. Inhibition of the janus kinase family increases extracellular signal-regulated kinase 1/2 phosphorylation and causes endoreduplication. Cancer Res 2006; 66:9083 - 9089
- David M, Petricoin E 3rd, Benjamin C, Pine R, Weber MJ, Larner AC. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science 1995; 269:1721 - 1723
- Caraglia M, Vitale G, Marra M, Budillon A, Tagliaferri P, Abbruzzese A. Alpha-interferon and its effects on signalling pathways within cells. Curr Protein Pept Sci 2004; 5:475 - 485
- Pircher TJ, Petersen H, Gustafsson JA, Haldosen LA. Extracellular signal-regulated kinase (ERK) interacts with signal transducer and activator of transcription (STAT) 5a. Mol Endocrinol 1999; 13:555 - 565
- Jain N, Zhang T, Fong SL, Lim CP, Cao X. Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK). Oncogene 1998; 17:3157 - 3167
- Sengupta TK, Talbot ES, Scherle PA, Ivashkiv LB. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci USA 1998; 95:11107 - 11112
- Loucks FA, Le SS, Zimmermann AK, Ryan KR, Barth H, Aktories K, et al. Rho family GTPase inhibition reveals opposing effects of mitogen-activated protein kinase kinase/extracellular signal-regulated kinase and Janus kinase/signal transducer and activator of transcription signaling cascades on neuronal survival. J Neurochem 2006; 97:957 - 967
- Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of Raf as a result of recruitment to the plasma membrane. Science 1994; 264:1463 - 1467
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 1996; 84:889 - 897
- Davis RJ. MAPKs: new JNK expands the group. Trends Biochem Sci 1994; 19:470 - 473
- Laird AD, Taylor SJ, Oberst M, Shalloway D. Raf-1 is activated during mitosis. J Biol Chem 1995; 270:26742 - 26745
- Wang S, Ghosh RN, Chellappan SP. Raf-1 physically interacts with Rb and regulates its function: a link between mitogenic signaling and cell cycle regulation. Mol Cell Biol 1998; 18:7487 - 7498
- Eves EM, Shapiro P, Naik K, Klein UR, Trakul N, Rosner MR. Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol Cell 2006; 23:561 - 574
- Greene LM, Campiani G, Lawler M, Williams DC, Zisterer DM. BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1,5-benzoxapines. Mol Pharm 2008; 73:419 - 430
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ. Kinetochore localization and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci 2001; 114:4385 - 4395
- Choi E, Choe H, Min J, Choi JY, Kim J, Lee H. BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J 2009; 28:2077 - 2089
- Oikawa T, Okuda M, Ma Z, Goorha R, Tsujimoti H, Inokuma H, et al. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol 2005; 25:4046 - 4061
- Matsumura S, Toyoshima F, Nishida E. Polo-like kinase 1 facilitates chromosome alignment during prommetaphase through BubR1. J Biol Chem 2007; 282:15217 - 15227
- Morrow CJ, Tighe A, Johnson VL, Scott MIF, Ditchfield C, Taylor SS. Bub1 and aurora B cooperate to maintain BubR1-mediated inhibition of APC/C Cdc20. J Cell Sci 2005; 118:3639 - 3652
- Wang Q, Liu T, Fang Y, Xie S, Huang X, Mahmood R, et al. BubR1 deficiency results in abnormal megakaryopoiesis. Blood 2004; 103:1278 - 1285
- Cha H, Shapiro P. Tyrosine-phosphorylated extracellular signal—regulated kinase associates with the Golgi complex during G2/M phase of the cell cycle: evidence for regulation of Golgi structure. J Cell Biol 2001; 153:1355 - 1367
- Smith J, Buncaciu RP, Reiterer G, Coder D, George T, Asaly M, et al. Retinoic acid induces nuclear accumulation of Raf1 during differentiation of HL-60 cells. Exp Cell Res 2009; 315:2241 - 2248
- Yang YL, Duan Q, Guo TB, Wang XX, Ruan Q, Xu GT, et al. BubR1 deficiency results in enhanced activation of MEK and ERKs upon microtubule stresses. Cell Prolif 2007; 40:397 - 410
- Jorquera R, Tanguay RM. Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum Mol Genet 2001; 15:1741 - 1752
- Prochownik EV. Functional and physical communication between oncoproteins and tumor suppressors. Cell Mol Life Sci 2005; 62:2438 - 2459