Abstract
During S-phase of the cell cycle, chromosomal DNA is replicated according to a complex replication timing program, with megabase-sized domains replicating at different times. DNA fibre analysis reveals that clusters of adjacent replication origins fire near-synchronously. Analysis of replicating cells by light microscopy shows that DNA synthesis occurs in discrete foci or factories. The relationship between timing domains, origin clusters and replication foci is currently unclear. Recent work, using a hybrid Xenopus/hamster replication system, has shown that when CDK levels are manipulated during S-phase the activation of replication factories can be uncoupled from progression through the replication timing program. Here, we use data from this hybrid system to investigate potential relationships between timing domains, origin clusters and replication foci. We suggest that each timing domain typically comprises several replicon clusters, which are usually processed sequentially by replication factories. We discuss how replication might be regulated at different levels to create this complex organisation and the potential involvement of CDKs in this process.
To ensure genetic stability the entire genome must be accurately duplicated prior to cell division. The demand for absolute fidelity requires that genome duplication is precisely regulated and furthermore, that it is coordinated with other cellular processes such as gene transcription and chromatin modification, in addition to the division process per se. Chromosomal DNA replication must therefore be highly regulated at all stages from replication initiation through to the final proof reading and repair of the duplicated genome.
DNA replication initiates at multiple sites distributed throughout each eukaryotic genome. Many thousands of replication origins are activated at distinct and reproducible times throughout S-phase according to a tightly controlled temporal program.Citation1–Citation3 In general, transcriptionally active euchromatin replicates early in S-phase, whereas transcriptionally inactive heterochromatin replicates late. Individual chromosome domains that replicate coincidentally are localized in specific regions of the nucleus.Citation1–Citation3 This can be seen as sequential patterns of DNA replication during the course of S-phase (). Although not absolutely discrete, five phases of replication (Types I, II, III, IV and V) can be distinguished: bulk nucleoplasmic, peripheral and perinuclear-perinucleolar localization precede replication in first small and then large, intranuclear patches.Citation4–Citation6 The ‘replication timing program’ thus coordinates replication in both a spatial and a temporal manner, and is well conserved amongst metazoans.Citation2
At the end of mitosis there is a major re-organization of the genome as chromosomes decondense and coalesce to form the interphase nucleus ().Citation7 Shortly after chromosome decondensation occurs, specific chromosomal domains move to occupy certain positions within the nucleus (). This movement of subchromosomal domains to appropriate positions within the nucleus in early G1 coincides with the establishment of the replication timing program, and is called the Timing Decision Point.Citation5 If nuclei are driven into S-phase prior to this, chromosomal domains are replicated in no particular order. The positioning of subchromosomal domains correlates broadly with local chromatin organization and the underlying transcriptional activity.Citation1–Citation3
Constitutively early replicating euchromatic domains take up a generally more open chromatin conformation and localize to the nuclear interior. The underlying DNA is GC rich, has a high gene density and is typically marked by hyperacetylated histones H3 and H4. A high percentage of genes within these subdomains are actively transcribed. In contrast, later replicating heterochromatic domains are localized towards the nuclear periphery or in large intranuclear substructures, such as is found around the nucleoli. These regions, which are marked by hypoacetylation of histones H3 and H4, form a more constrained, less open, chromatin structure and contain a lower percentage of actively transcribed genes. Heterochromatin tends to be both GC poor and have a low gene density.Citation1–Citation3 Changes in the transcriptional status of genes from GC poor regions during development are coupled to changes in nuclear position and replication timing such that active transcription correlates with repositioning towards the nuclear interior and earlier replication timing.Citation8–Citation10 Replication timing correlates better with the transcriptional competence of the chromosome domain than with the activity status of any individual gene.Citation3,Citation11 The relationship between gene transcription and replication timing is reciprocal. Whereas chromatin formed from DNA replicating early in S-phase is characterized by the deposition of actively acetylated histones H3 and H4, the nuclear environment appears to be altered during the course of S-phase such that later replicating DNA is packaged with deacetylated histones.Citation12,Citation13 This provides a potential mechanism to facilitate the propagation of the local chromatin environment during DNA replication.
To further investigate the execution of the replication timing program during S-phase we used a hybrid experimental system developed by Gilbert and colleagues in which the replication of mammalian Chinese Hamster Ovary (CHO) cells nuclei is driven by incubation in Xenopus laevis (frog) egg extracts.Citation14,Citation15 Nuclei prepared from CHO cells in mid G1, beyond the Timing Decision Point, are programed to replicate according to the in vivo timing program.Citation5 At 20 minutes, the length of S-phase in the early Xenopus embryo is significantly shorter than that of the ∼12 hour S-phase of CHO cells. When incubated in Xenopus egg extracts, the CHO nuclei undergo a significantly accelerated timing program lasting ∼2 hours.Citation14,Citation15 Replication labeling patterns, resembling those observed in vivo, develop sequentially during the in vitro incubation (). However, in addition to the more discrete patterns discerned in vivo, combined patterns (Types I/II, II/III, III/IV, IV/V) were also observed. This suggests that in vitro, later stages of the timing program can start while previous stages are still ongoing. Although most nuclei proceed to late replication patterns, overall DNA replication is inefficient, averaging less than 40% of template DNA replicated.Citation14,Citation15 Despite the ∼6-fold reduction in S-phase length in vitro, the amount of DNA synthesis associated with each replication pattern is proportionally similar to the time spent replicating each pattern in vivo.Citation15 Taken together these results suggested that the replication program can progress to later timing stages before completing earlier steps. This means that replication initiation and the replication timing program are not tightly coordinated and so may be differentially regulated.
The Role of Cdks in S-Phase Progression
During late mitosis, and early G1, each replication origin is first loaded with Mcm2-7 to form a prereplicative complex that licenses DNA for a single initiation event in the following S-phase ().Citation16,Citation17 During S-phase each licensed origin is then activated by Cdks and Dbf4-dependent kinases (Ddks) to induce initiation. The combined action of these two kinases promotes the recruitment of replisome proteins to the origin and the establishment of active replication forks. The number of origins licensed in each cell cycle is in significant excess over the number of origins actually used during S-phase.Citation18–Citation20 Otherwise dormant origins can be activated when replication fork progression is inhibited, providing a means to maintain DNA replication rates under conditions of replicative stress.Citation21–Citation23 In an unperturbed S-phase dormant origins are usually passively replicated, and inactivated, by forks initiated at an adjacent origin. The selection of origins for activation in the coming S-phase, the ‘origin decision point’ occurs during G1 phase after the establishment of replication timing ().Citation24,Citation25 It is unclear what makes some origins efficient and some origins inefficient (usually dormant). One possibility is that efficient origins are preferential targets for the initiating activities of Cdks and Ddks.
We therefore investigated the effect of modulating Cdk activity in the egg extract, using the chemical Cdk inhibitor roscovitine and recombinant cyclin A.Citation15 If strictly coupled, both the rate of replication and progression through the timing program would respond to alteration of Cdk activity. Conversely, if progression through the timing program was independent of both then no effect on timing would be observed.
Addition of increasing concentrations of roscovitine to egg extract, over a range of 1 to 100 µM, reduced Cdk activity to near background levels. Consistent with previous reports the rate of DNA replication reduced with similar kinetics, suggesting that Cdk activity is rate limiting for replication.Citation26–Citation29 Whereas roscovitine concentrations of 1–10 µM reduced the rate of DNA replication by up to ∼50%, the timing program was little affected. Only at roscovitine concentrations of 30 µM and above, concentrations at which the rates of DNA replication were reduced to 30% or less, was the timing program retarded. Conversely, supplementing S-phase Cdk activity by addition of cyclin A to egg extract, stimulated DNA replication while only moderately accelerating the timing program. These results suggest that progression of the timing program is dependent, either directly or indirectly, on Cdk activity. Further, the differential responses of replication rate and replication timing to moderately reduced Cdk activity suggests that timing progression and replication initiation are differentially regulated by Cdks, allowing progression to later stages of the replication program while earlier stages are incomplete (). This conclusion was further demonstrated when replication timing and total DNA synthesis were quantified in individual nuclei.Citation15 Whereas roscovitine significantly lowered the total amount of DNA synthesis associated with each pattern, cyclin A addition increased it. This directly demonstrates a decoupling between replication rate and progression through the replication timing program.
Replicon Clusters, Replication Factories and Subchromosomal Timing Domains
In typical mammalian tissue culture cells, the DNA replicated from an individual origin, the replicon, typically ranges in size from 20 to 450 kb, with most being between 50 and 150 kb.Citation30–Citation33 Most replicons are organized into clusters of synchronously initiating adjacent origins. Replicon clusters, which are composed of two to ten proximal origins, typically encompass less than 1 Mb of DNA and fully replicate in 45 to 60 minutes. Genomic analysis shows that relatively large domains of contiguous DNA are replicated at characteristic times during S-phase.Citation10,Citation34–Citation39 These subchromosomal domains vary between different cell types and are subject to developmental regulation. For example, embryonic stem cells have a mean timing domain size of 1.5 Mbp, while in neural precursor cells the median size is 2.5 Mbp. These sizes suggest that timing domains typically comprise more than one adjacent replicon clusters.Citation10
Clusters of origins can be visualized in living cells as discrete subnuclear foci and these remain stable through multiple cell divisions.Citation5,Citation32,Citation40–Citation42 Foci of ongoing replication are enriched for replication fork proteins, marking factories of DNA synthesis.Citation6,Citation43–Citation46 It remains to be determined whether foci form as a physical factory in which proteins associated with replication forks emanating from adjacent origins are organized as a superstructure or whether the colocalization is dependent on the underlying organization of chromosomal DNA. Over a period of 1 to 2 hours fork proteins are observed at sites increasingly distal to the initial focus of replication, conceivably relocalising to adjacent replicons or replicon clusters.Citation42,Citation45 The precise relationship between replicon clusters, timing domains and replication foci remains unclear. We also have very little understanding of how their activation is regulated during S-phase.
To address the regulation of these structures, we investigated whether altering Cdk levels in vitro affected the rate of initiation within a factory or the rate of activation of new factories.Citation15 Treatment of extracts with up to 10 µM roscovitine reduced the total number of replication foci while leaving their intensity relatively unchanged (). Consistent with this, 10 µM roscovitine did not significantly change either replication fork speed or the density of forks within active replicon clusters. These low concentrations of roscovitine had only very limited effects on the timing program. At higher concentrations of roscovitine, which more strongly inhibited DNA replication and passage through the timing program, both foci number and intensity were significantly affected. Conversely, stimulating DNA replication with cyclin A increased the number of active foci (). Modest changes in Cdk activity therefore preferentially alters the activation of new replication factories, leaving initiation within clusters relatively unaffected, while the threshold of Cdk activity required for replication factory activation is greater than that required to drive progression through the timing program ().
Regulation of Factories and Origin Clusters
Compared to the initiation of new replicons within an existing cluster or progression through the timing program, the activation of a new replication factory is highly sensitive to CDK activity. This may reflect the requirement for an additional Cdk substrate, distinct from the substrates required for individual origins, which acts to facilitate the initiation of all origins within a cluster or domain, thereby functioning as a factory activator. Alternatively, there might be a requirement for high Cdk activity to activate the first origin in a cluster, the firing of which then propagates a change throughout the cluster or domain to facilitate initiation at other origins. This “founder effect” can give the observed CDK-sensitivity of factory number even if other non-CDK activities independently influence factory number. In either interpretation, there has to be an effect that propagates throughout a cluster or domain to activate it for replication. This could plausibly reflect a change to the chromatin state or higher-order chromatin structure. One possibility is that CDKs regulate chromatin remodelling complexes that affect the access of the replication machinery. Nucleosome remodelling and reassembly is a key component of S-phase progression.Citation47 The SNF2H chromatin remodelling complex has been shown to facilitate the access of replication factors to the replication origin of the Epstein-Barr virus,Citation48 while a SNF2H subcomplex has been shown to be required for efficient replication of heterochromatin in mouse and human cells.Citation49 A role for CDKs in this process is also suggested by the observation that the initiation protein Cdc45 recruits Cdk2 to specific chromosome sites and promotes CDK-dependent chromatin decondensation.Citation50
In the hybrid system, analysis of total nuclear foci number reveals that ∼2,400 replication foci are active during early S-phase when type II replication patterns predominate. This is 2–3 times more active foci than are observed in typical replicating somatic cells which is possibly a consequence of higher CDK levels in Xenopus egg extracts as compared to CHO cells. The increased number of active replication factories provides a potential explanation for the >2-fold increased replication rate when CHO nuclei are incubated in Xenopus egg extract.
The peak nuclear replication rate seen when CHO nuclei replicate in vitro is ∼20–30 Mbp/min,Citation15 and with a fork rate of 10 nt/sec, this suggests that each nucleus contains 30–50,000 active forks. Therefore each of the 2,400 foci has on average 8–12 replication forks. Inhibition of initiation during early S-phase in the hybrid system reduced replication rates to 16% within 20–30 min, and with forks being present every ∼10 kb within a cluster,Citation15 this is consistent with each cluster containing ∼12 forks (). Taken together, these results are consistent with a model where each factory is processing a single cluster of replicons spanning ∼100 kb. This cluster size is significantly smaller than the 1.5–2.5 Mbp mean size of replication timing domains, and consistent with previous reports, suggests that a timing domain typically consists of more than one replicon cluster ().Citation10 In somatic tissue culture cells, the different replicon clusters comprising a single timing domain are likely activated sequentially, possibly leading to the appearance of factories at adjacent sites, the so-called domino model ().Citation42,Citation45 In that case, the increased number of replication factories seen when CHO nuclei replicate in Xenopus extracts might be due to the simultaneous activation of different clusters within individual timing domains ().
Although other explanations are possible, the replication parameters observed in the hybrid system are consistent with this interpretation. The initiation events associated with Type II DNA in the hybrid system are largely confined to a window of ∼20 mins.Citation15 This is similar to the time required to complete the replication of a cluster, meaning that there can be no more than two waves of cluster activation in this time. If ∼40% of Type II DNA is replicated in the hybrid system, and this represents no more than two waves of cluster activation, then the density of active 100 kb clusters within a Type II domain must be 2 or more per Mbp. With a 1.5–2.5 Mbp expected size range of timing domains, we would expect that most active timing domains would have more than one cluster active at any one time.
Regulation of Progression through the Timing Program
Whereas the establishment of the replication timing program in this hybrid system is a property of the CHO nuclei, the execution of the program requires interaction between the nuclei and the Xenopus egg extract. One possibility is that execution could be regulated in cis, at the chromatin level, so that progression from one stage of the program to another only occurs when all the initiation events associated with the earlier timing stage have been completed. As such the activation or perhaps the completion, of replication in one factory would facilitate the activation, in cis, of an adjacent replication factory in a then later replicating subchromosomal domain. This strict coupling is outlined schematically in . However, this does not appear to be the case in the hybrid system. CHO nuclei replicating in vitro progress to late replication patterns with less than half the template DNA replicated.Citation5,Citation15 The appearance of combined replication patterns (I/II, II/III, etc.,) and the overlap in the initiation times associated with different patterns also argue against this sort of strict coupling. When replication was inhibited in CHO cells in the presence of checkpoint kinase inhibitors, the activation of late replicating domains occurred on schedule,Citation51 which also argues against strict coupling between DNA replication and progression through the timing program. Further, the observed replication timing profiles of proliferating somatic cells does not support the idea of replication being strictly dependent on cis-acting effects propagating along chromosomes.Citation10,Citation34–Citation37,Citation52 The execution of the timing program is therefore unlikely to be mediated merely in cis but requires an activity that functions independent of preceding replication.
An alternative possibility is that execution is regulated in trans, for example, by increasing levels of Cdk activity in the extract. Following this interpretation, initiation at later-firing origins would require higher Cdk levels that are only achieved later in S-phaseCitation53 (). However, models of this type cannot account for the decoupling seen when we artificially raised and lowered CDK levels, as this should cause both initiation rate, factory numbers and progression through the timing program to change together. Instead, our results suggest that CDKs drive DNA replication in several distinct ways. In addition to being required for individual origins to fire (), CDKs appear to be separately required for activation of individual replication factories and to drive progression through the replication timing program.
What could be the substrate that regulates progression through the timing program? Late replicating DNA is typically organized into heterochromatin.Citation1–Citation3 This is characterized by a less open chromatin structure that induces volume exclusion and limits protein diffusion.Citation54 Akin to the activation of gene transcription, it will be necessary to overcome any restrictions that this environment imposes to facilitate the initiation of DNA replication in these compartments. The replication timing program may then reflect the progressive, and perhaps successive, relaxation of distinct heterochromatic states. A timing factor or factors, perhaps specific to discrete heterochromatic domains, whose activation requires only a low threshold of Cdk activity, might therefore function, in trans, over time, to reverse these restrictive environments. Although providing a barrier to domain activation, the compact nature of heterochromatin may in fact facilitate this relaxation once such a timing factor is activated, as the molecular crowding seen in these compartments serves to enhance the affinity of factors for the chromatin.Citation54 Conversely, the Cdk dependent inactivation or relocalization, of activities required to maintain the inhibitory state could also serve to alleviate the restriction. Throughout the course of S-phase then discrete regions of the genome might be made accessible to the replication machinery. Candidate timing factors include both chromatin remodelling and structural proteins. SNF2H, Histone H1, HMG-1 and HP1, have each been shown to affect the replication timing of specific loci.Citation49,Citation55–Citation57 In vivo the activation of a timing factor(s) might occur over time, possibly co-ordinated with cell cycle status, rather than with replication progression per se, in a changing nuclear environment. In the hybrid system, however, progression through the timing program is accelerated, so that late replicating chromatin becomes available for replication before replication of early DNA is complete, with the activation of new clusters or replication factories being rate-limiting. This could explain the occurrence of the composite patterns and other features of replication observed in the hybrid system.
Figures and Tables
Figure 1 The replication timing program. (A) CHO cells, previously synchronized at the G1/S border and released into S-phase for various times, were pulse labelled with BrdU for 5 min and stained with anti-BrdU antibodies to visualize patterns of DNA replication. Shown are characteristic examples of each of the 5 labeling patterns (Types I–V). Reproduced from Dimitrova & Gilbert 1999. (B) The replication cycle—the establishment and execution of replication timing in the cell cycle. The innermost wheel marks the phases of the cell cycle and the outermost wheel the key stages of DNA replication, where the periods of licensing competence and inhibition are shown in yellow and red respectively and the execution of the replication timing program during S-phase is shown in green. Key nuclear transitions are represented diagrammatically in the central portion. During mitosis, paired sister chromatids aligned on the metaphase plate are separated to opposite poles of the dividing cells upon entry into anaphase. Late mitotic chromosome decondensation then readies DNA for emergence into G1. In early G1, after nuclear envelope reformation (continuous black line), regions of euchromatin, shown in red and heterochromatin, shown in blue, initially randomly dispersed, move to occupy specific domains within the nucleus. During S-phase, replication foci, shown in green, are activated in accordance with the execution of the timing program. Post-replicative nuclei pass through G2 into mitosis, where chromosome condensation and nuclear envelope breakdown (dashed black line) ready the cell for sister chromatid separation and cell division. The execution of the timing decision point and the origin decision point are marked.
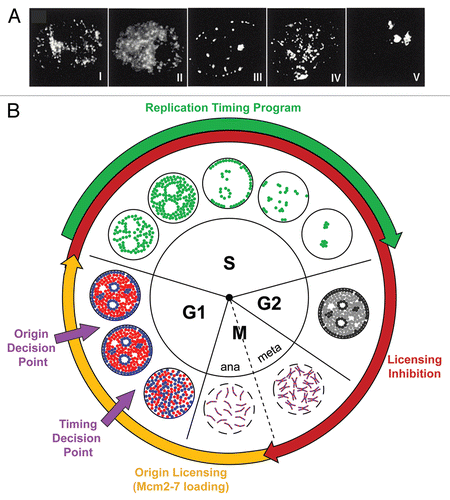
Figure 2 Progression of CHO nuclei through the replication timing program in Xenopus egg extract. CHO nuclei, incubated in Xenopus egg extracts, were pulse labelled for 5 min, at various times, with Cy3-dUTP to directly visualize patterns of DNA replication. Shown are characteristic examples of each of the 9 labeling patterns, the 5 distinct patterns (Types I–V) and the 4 overlapping patterns (Types I/II, II/III, III/IV, IV/V). Reproduced from Thomson et al., 2010.
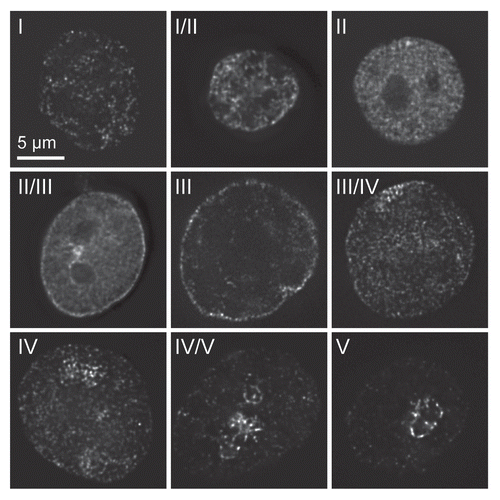
Figure 3 Decoupling of DNA replication and the replication timing program. CHO nuclei were incubated in Xenopus egg extracts ±10 µM roscovitine (A) or 1 pM cyclin A (B). At various times, aliquots were pulse labelled with Cy3-dUTP to assess the proportion of different replication patterns. At the same time DNA synthesis was measured in extracts supplemented with α-[32P]dATP by TCA precipitation and scintillation counting. The replication pattern at different times is plotted against total DNA synthesis. Reproduced from Thomson et al., 2010.
![Figure 3 Decoupling of DNA replication and the replication timing program. CHO nuclei were incubated in Xenopus egg extracts ±10 µM roscovitine (A) or 1 pM cyclin A (B). At various times, aliquots were pulse labelled with Cy3-dUTP to assess the proportion of different replication patterns. At the same time DNA synthesis was measured in extracts supplemented with α-[32P]dATP by TCA precipitation and scintillation counting. The replication pattern at different times is plotted against total DNA synthesis. Reproduced from Thomson et al., 2010.](/cms/asset/842db161-4cf9-4ebc-a2da-3c46b523f686/kccy_a_10912644_f0003.gif)
Figure 4 Activation of replication foci depends on CDK levels. (A) CHO nuclei, were incubated in Xenopus egg extracts for 50 min ± 10 µM roscovitine or 3 pM cyclin A and then pulse labelled for 5 min with Cy3-dUTP (red) and then isolated, stained with DAPI (blue) and visualized. Representative images for each condition are shown. Reproduced from Thomson et al., 2010. (B) CDK sensitivity of DNA replication control. Cartoon showing three different levels of S phase control. The upper level shows progression between two different stages of the timing program for a single nucleus, where the small green dots represent replication factories and the black circle the nuclear envelope. The middle level represents the firing of replication origins (pink dots) in a newly activated replication factory next to an existing factory (large green dot). The lower level shows initiation of a new replication origin on a strand of DNA in an active replicon cluster within a replication factory, where the pink circle represents the new origin and the green line the DNA.
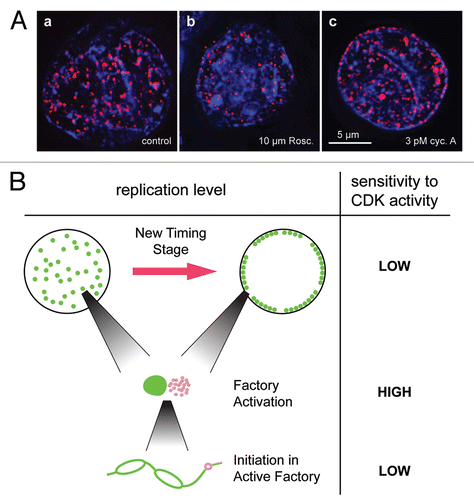
Figure 5 The organization of replicon clusters. (A) CHO nuclei were incubated in Xenopus egg extracts supplemented with α-[32P]dATP. At 40 min an aliquot was supplemented with 1 mM roscovitine to block further initiation events. At various times throughout the length of the incubation total DNA synthesis was measured by TCA precipitation and scintillation counting. Reproduced from Thomson et al., 2010. (B) Replication fork fusion within a replicon cluster reduces replication rate. Cartoon depicting the effect of replication fork fusion on reducing replication rate within a replicon cluster, where DNA is represented by a black line and open figures represent ‘replication bubbles’, the ongoing synthesis of DNA from a pair of replication forks moving in opposite directions. Replication fork number and density and replication fork rate and termination time, within a cluster, are indicated.
![Figure 5 The organization of replicon clusters. (A) CHO nuclei were incubated in Xenopus egg extracts supplemented with α-[32P]dATP. At 40 min an aliquot was supplemented with 1 mM roscovitine to block further initiation events. At various times throughout the length of the incubation total DNA synthesis was measured by TCA precipitation and scintillation counting. Reproduced from Thomson et al., 2010. (B) Replication fork fusion within a replicon cluster reduces replication rate. Cartoon depicting the effect of replication fork fusion on reducing replication rate within a replicon cluster, where DNA is represented by a black line and open figures represent ‘replication bubbles’, the ongoing synthesis of DNA from a pair of replication forks moving in opposite directions. Replication fork number and density and replication fork rate and termination time, within a cluster, are indicated.](/cms/asset/3ac151d8-4ef7-4e38-9a65-c4eda65cbfbd/kccy_a_10912644_f0005.gif)
Figure 6 Replication timing domains are comprised of multiple replicon clusters. (A) Genomic replication timing profile of 158.4–162.7 Mbp of mouse chromosome 2, in neural precursor (NPC/ASd6) cells.Citation10,Citation58 A broad region of DNA with approximately coordinately timed replication initiation constitutes a replication timing domain. Within the fine detail of each timing domain smaller divisions of replication timing, perhaps representing individual replicon clusters, can be discerned. Data prepared using www.replicationdomain.org. (B and C) Cartoon showing hypothetical replicon cluster activation within a replication timing domain similar to the one shown in (A), in (B) CHO cells and (C) the hybrid system. Currently active clusters, those in which the internal forks are still active and have not yet fused, are depicted in red. Completed replicon clusters, those in which the internal forks have fused but the outermost forks remain active, are depicted in blue. Unreplicated DNA is represented by the black line. The size of a cluster in the 2 systems is scaled relative to their observed length.
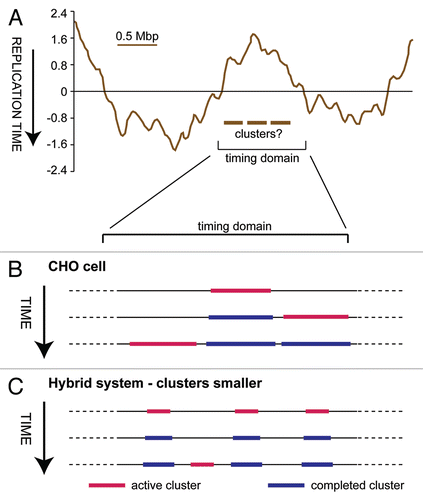
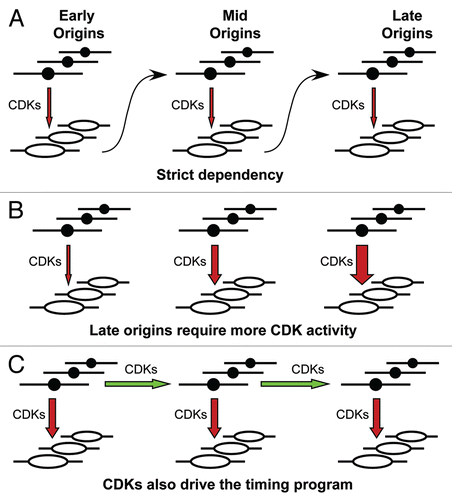
Figure 7 Different models for how the replication timing program might operate. Groups of replication origins, represented as black circles, on early-, mid- and late-replicating DNA are shown in each panel. CDKs acting on these origins to promote initiation and turn them into replication bubbles, are depicted as red vertical arrows. (A) Initiation of origins in one timing stage is dependent on initiation of the previous stage having been completed. (B) Initiation of later origins requires increased CDK activity, so the replication timing program is driven by rising CDK levels. (C) CDK activity is required not only to promote origin activation (vertical arrows), but also to drive the replication timing program (horizontal arrows), so that later origins become competent for initiation.
Acknowledgements
Thanks to Dave Gilbert for discussions relating to this review and to Alistair Thomson for the original version of Figure 1B. This work was supported by Cancer Research UK (grants C303/A3135, C303/A5434, C303/A7399) and the Biotechnology and Biological Sciences Research Council (grants S10310, BB/H013024/1).
References
- Goren A, Cedar H. Replicating by the clock. Nat Rev Mol Cell Biol 2003; 4:4 - 6
- Zink D. The temporal program of DNA replication: new insights into old questions. Chromosoma 2006; 115:273 - 287
- Pope BD, Hiratani I, Gilbert DM. Domain-wide regulation of DNA replication timing during mammalian development. Chromosome Res 2010; 18:127 - 136
- O'Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol 1992; 116:1095 - 1110
- Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell 1999; 4:983 - 993
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, et al. Dynamics of DNA replication factories in living cells. J Cell Biol 2000; 149:271 - 280
- Hetzer MW, Walther TC, Mattaj IW. Pushing the envelope: structure, function and dynamics of the nuclear periphery. Annu Rev Cell Dev Biol 2005; 21:347 - 380
- Perry P, Sauer S, Billon N, Richardson WD, Spivakov M, Warnes G, et al. A dynamic switch in the replication timing of key regulator genes in embryonic stem cells upon neural induction. Cell Cycle 2004; 3:1645 - 1650
- Hiratani I, Leskovar A, Gilbert DM. Differentiation-induced replication-timing changes are restricted to AT-rich/long interspersed nuclear element (LINE)-rich isochores. Proc Natl Acad Sci USA 2004; 101:16861 - 16866
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 2008; 6:245
- MacAlpine DM, Rodriguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev 2004; 18:3094 - 3105
- Zhang J, Xu F, Hashimshony T, Keshet I, Cedar H. Establishment of transcriptional competence in early and late S phase. Nature 2002; 420:198 - 202
- Lande-Diner L, Zhang J, Cedar H. Shifts in replication timing actively affect histone acetylation during nucleosome reassembly. Mol Cell 2009; 34:767 - 774
- Gilbert DM, Miyazawa H, DePamphilis ML. Site-specific initiation of DNA replication in Xenopus egg extract requires nuclear structure. Mol Cell Biol 1995; 15:2942 - 2954
- Thomson AM, Gillespie PJ, Blow JJ. Replication factory activation can be decoupled from the replication timing program by modulating Cdk levels. J Cell Biol 2010; 188:209 - 221
- Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol 2005; 6:476 - 486
- Nishitani H, Lygerou Z. DNA replication licensing. Front Biosci 2004; 9:2115 - 2132
- Burkhart R, Schulte D, Hu D, Musahl C, Gohring F, Knippers R. Interactions of human nuclear proteins P1Mcm3 and P1Cdc46. Eur J Biochem 1995; 228:431 - 438
- Lei M, Kawasaki Y, Tye BK. Physical interactions among Mcm proteins and effects of Mcm dosage on DNA replication in Saccharomyces cerevisiae. Mol Cell Biol 1996; 16:5081 - 5090
- Mahbubani HM, Chong JP, Chevalier S, Thömmes P, Blow JJ. Cell cycle regulation of the replication licensing system: involvement of a Cdk-dependent inhibitor. J Cell Biol 1997; 136:125 - 135
- Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 2007; 21:3331 - 3341
- Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, et al. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 2006; 173:673 - 683
- Ibarra A, Schwob E, Mendez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci USA 2008; 105:8956 - 8961
- Wu JR, Gilbert DM. A distinct G1 step required to specify the Chinese hamster DHFR replication origin. Science 1996; 271:1270 - 1272
- Li F, Chen J, Solessio E, Gilbert DM. Spatial distribution and specification of mammalian replication origins during G1 phase. J Cell Biol 2003; 161:257 - 266
- Strausfeld UP, Howell M, Rempel R, Maller JL, Hunt T, Blow JJ. Cip1 blocks the initiation of DNA replication in Xenopus extracts by inhibition of cyclin-dependent kinases. Curr Biol 1994; 4:876 - 883
- Strausfeld UP, Howell M, Descombes P, Chevalier S, Rempel RE, Adamczewski J, et al. Both cyclin A and cyclin E have S-phase promoting (SPF) activity in Xenopus egg extracts. J Cell Sci 1996; 109:1555 - 1563
- Moore JD, Kornbluth S, Hunt T. Identification of the nuclear localization signal in Xenopus cyclin E and analysis of its role in replication and mitosis. Mol Biol Cell 2002; 13:4388 - 4400
- Luciani MG, Oehlmann M, Blow JJ. Characterization of a novel ATR-dependent, Chk1-independent, intra-S-phase checkpoint that suppresses initiation of replication in Xenopus. J Cell Sci 2004; 117:6019 - 6030
- Edenberg HJ, Huberman JA. Eukaryotic chromosome replication. Annu Rev Genet 1975; 9:245 - 284
- Yurov YB, Liapunova NA. The units of DNA replication in the mammalian chromosomes: evidence for a large size of replication units. Chromosoma 1977; 60:253 - 267
- Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 1998; 140:1285 - 1295
- Berezney R, Dubey DD, Huberman JA. Heterogeneity of eukaryotic replicons, replicon clusters and replication foci. Chromosoma 2000; 108:471 - 484
- Watanabe Y, Fujiyama A, Ichiba Y, Hattori M, Yada T, Sakaki Y, et al. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: disease-related genes in timing-switch regions. Hum Mol Genet 2002; 11:13 - 21
- White EJ, Emanuelsson O, Scalzo D, Royce T, Kosak S, Oakeley EJ, et al. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc Natl Acad Sci USA 2004; 101:17771 - 17776
- Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, et al. Replication timing of the human genome. Hum Mol Genet 2004; 13:191 - 202
- Desprat R, Thierry-Mieg D, Lailler N, Lajugie J, Schildkraut C, Thierry-Mieg J, et al. Predictable dynamic program of timing of DNA replication in human cells. Genome Res 2009; 19:2288 - 2299
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 2010;
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 2010; 20:155 - 169
- Ma H, Samarabandu J, Devdhar RS, Acharya R, Cheng PC, Meng C, et al. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J Cell Biol 1998; 143:1415 - 1425
- Zink D, Bornfleth H, Visser A, Cremer C, Cremer T. Organization of early and late replicating DNA in human chromosome territories. Exp Cell Res 1999; 247:176 - 188
- Sadoni N, Cardoso MC, Stelzer EH, Leonhardt H, Zink D. Stable chromosomal units determine the spatial and temporal organization of DNA replication. J Cell Sci 2004; 117:5353 - 5365
- Nakamura H, Morita T, Sato C. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res 1986; 165:291 - 297
- Somanathan S, Suchyna TM, Siegel AJ, Berezney R. Targeting of PCNA to sites of DNA replication in the mammalian cell nucleus. J Cell Biochem 2001; 81:56 - 67
- Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol Cell 2002; 10:1355 - 1365
- Kitamura E, Blow JJ, Tanaka TU. Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell 2006; 125:1297 - 1308
- Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell 2007; 128:721 - 733
- Zhou J, Chau CM, Deng Z, Shiekhattar R, Spindler MP, Schepers A, et al. Cell cycle regulation of chromatin at an origin of DNA replication. EMBO J 2005; 24:1406 - 1417
- Collins N, Poot RA, Kukimoto I, Garcia-Jimenez C, Dellaire G, Varga-Weisz PD. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet 2002; 32:627 - 632
- Alexandrow MG, Hamlin JL. Chromatin decondensation in S-phase involves recruitment of Cdk2 by Cdc45 and histone H1 phosphorylation. J Cell Biol 2005; 168:875 - 886
- Dimitrova DS, Gilbert DM. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol 2000; 2:686 - 694
- Karnani N, Taylor C, Malhotra A, Dutta A. Pan-S replication patterns and chromosomal domains defined by genome-tiling arrays of ENCODE genomic areas. Genome Res 2007; 17:865 - 876
- Erlandsson F, Linnman C, Ekholm S, Bengtsson E, Zetterberg A. A detailed analysis of cyclin A accumulation at the G(1)/S border in normal and transformed cells. Exp Cell Res 2000; 259:86 - 95
- Bancaud A, Huet S, Daigle N, Mozziconacci J, Beaudouin J, Ellenberg J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J 2009; 28:3785 - 3798
- Flickinger R. Replication timing and cell differentiation. Differentiation 2001; 69:18 - 26
- Thiriet C, Hayes JJ. Linker histone phosphorylation regulates global timing of replication origin firing. J Biol Chem 2009; 284:2823 - 2829
- Hayashi MT, Takahashi TS, Nakagawa T, Nakayama J, Masukata H. The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat Cell Biol 2009; 11:357 - 362
- Weddington N, Stuy A, Hiratani I, Ryba T, Yokochi T, Gilbert DM. ReplicationDomain: a visualization tool and comparative database for genome-wide replication timing data. BMC Bioinformatics 2008; 9:530