Abstract
The DNA damage checkpoint maintains genome stability by arresting the cell cycle and promoting DNA repair under genotoxic stress. Cells must downregulate the checkpoint signaling pathways in order to resume cell division after completing DNA repair. While the mechanisms of checkpoint activation have been well-characterized, the process of checkpoint recovery, and the signals regulating it, has only recently been investigated. We have identified a new role for the Ras signaling pathway as a regulator of DNA damage checkpoint recovery. Here we report that in budding yeast, deletion of the IRA1 and IRA2 genes encoding negative regulators of Ras prevents cellular recovery from a DNA damage induced arrest. The checkpoint kinase Rad53 is dephosphorylated in an IRA-deficient strain, indicating that recovery failure is not caused by constitutive checkpoint pathway activation. The ira1D ira2D recovery defect requires the checkpoint kinase Chk1 and the cAMP-dependent protein kinase (PKA) catalytic subunit Tpk2. Furthermore, PKA phosphorylation sites on the anaphase promoting complex specificity factor Cdc20 are required for the recovery defect, indicating a link between the recovery defect and PKA regulation of mitosis. This work identifies a new signaling pathway that can regulate DNA damage checkpoint recovery, and implicates the Ras signaling pathway as an important regulator of mitotic events.
Introduction
Cell cycle checkpoints are pathways that ensure orderly progression through cell division.Citation1–Citation3 These pathways delay or arrest the cell cycle in response to adverse conditions or a failure of critical cell cycle events. The DNA damage checkpoint arrests the cell cycle to prevent replication of damaged DNA and to prevent segregation of damaged chromosomes, which can lead to genomic instability or aneuploidy. The DNA damage checkpoint can be broken into several steps: initiation, where a DNA lesion is detected and the signaling pathways are activated; maintenance, where the checkpoint signal remains active as the DNA lesion persists; and recovery, where the checkpoint signal is attenuated once the lesion has been repaired. In order for a cell to successfully cope with a DNA damage event, these phases must be appropriately regulated. Therefore, it is important to understand the mechanisms controlling checkpoint recovery as well as those controlling initiation and maintenance.
The signaling pathways that respond to DNA damage are conserved throughout evolution.Citation4,Citation5 In the budding yeast Saccharomyces cerevisiae, several types of DNA damage cause cell cycle arrest prior to the onset of anaphase. In metaphase of mitosis, sister chromatids are held together by Scc1/cohesin. In an unperturbed cell cycle, anaphase begins when Esp1/separase cleaves Scc1/cohesin. Pds1/securin sequesters Esp1/separase in an inactive state and plays a key role in preventing anaphase onset in the setting of DNA damage. The ubiquitin ligase anaphase promoting complex in association with the specificity factor Cdc20 (APCCdc20) drives the metaphase-to-anaphase transition by promoting ubiquitination of Pds1/securin, targeting it for proteasomal degradation. This frees Esp1/separase, which cleaves Scc1/cohesin and allows sister chromatid segregation. When cells suffer genotoxic insult, DNA lesions are processed to a form that activates the upstream sensor kinase Mec1 (mammalian ATR). Mec1 phosphorylates and activates two effector kinases, Chk1 and Rad53 (in mammalian cells the effector kinases are Chk1 and Chk2). Chk1 restrains mitosis by phosphorylating Pds1, preventing its ubiquitination by the APC.Citation6–Citation8 Rad53 can re-enforce preanaphase arrest by preventing Pds1 association with the APC and by inhibiting the mitotic exit network and preventing the degradation of mitotic cyclins.Citation6,Citation9 Consequently, Chk1 and Rad53 form two parallel pathways that converge on the APC substrates to restrain mitosis when cells have damaged DNA.
The cAMP-dependent protein kinase (PKA) pathway can participate in the DNA damage checkpoint.Citation10 Deletion of the PKA catalytic subunit TPK2 worsens the checkpoint defect of a chk1Δ strain (measured by cell division in the presence of DNA damage), demonstrating a role for PKA in delaying the metaphase-to-anaphase transition in a checkpoint-defective strain. PKA restrains mitosis by phosphorylating Cdc20 and preventing APCCdc20-mediated degradation of Pds1 and Clb2.Citation10 PKA responds to intracellular cAMP levels, which are positively regulated by adenlyate cyclase and negatively regulated by two phosphodiesterases.Citation11 Ras1 and Ras2, homologs of the mammalian proto-oncogenic Ras family members, activate yeast adenylate cyclase in response to increased glucose availability.Citation12,Citation13 Two GTPase activating proteins, Ira1 and Ira2, negatively regulate Ras and thereby restrain Ras and PKA signaling.Citation14–Citation16 Consequently, deletion of one or both IRA genes leads to increased Ras activation, accumulation of intracellular cAMP and increased PKA activity.Citation11,Citation17
Several pathways are involved in DNA damage checkpoint signal attenuation, and a number of deletion mutants with DNA damage recovery defects have been described.Citation18 Various protein phosphatases are required for dephosphorylation and inactivation of Rad53 following transient DNA damage. Different phosphatases are required for recovery from different types of damage. Recovery from a double strand break requires the PP2C-like Ptc2, while recovery from the DNA replication stress requires the PP1-related Glc7.Citation19–Citation21 Recovery-defective phosphatase-deficient strains arrest with high levels of phosphorylated Rad53, indicating that the mitotic exit defect is caused by a failure in inactivating the checkpoint signaling cascade. The DNA damage checkpoint signal is also attenuated during adaptation, a process where yeast cells resume cell division despite persistent irreparable damage.Citation22,Citation23 Like recovery, adaptation requires checkpoint kinase dephosphorylation and deactivation.Citation21,Citation24,Citation25 A strain with deletion of the gene encoding the SUMO protease Ulp2 is able to dephosphorylate Rad53 but nonetheless fails to adapt to a doublestrand DNA break, indicating that both dephosphorylation and desumoylation have critical roles in downregulating checkpoint activity, although the critical sumoylated checkpoint substrates involved in the adaptation defect remain undefined.Citation26
Considering the role of PKA signaling in regulation of mitosis and the DNA damage checkpoint, we wondered if deletion of the yeast IRA genes would affect DNA damage checkpoint activation, maintenance, or recovery. This question has bearing on human disease: mutation in the human IRA homolog NF1 tumor suppressor gene causes neurofibromatosis type 1, which predisposes to nervous system tumors originating from Schwann cell precursors (OMIM #162200).Citation27–Citation29 Loss of NF1 in Schwann cells increases intracellular cAMP levels and leads to PKA-dependent phenotypes, with increased cell migration and invasion similar to yeast cells with loss of the IRA genes.Citation30–Citation32 Individuals with NF1 are predisposed to radiation-induced neoplasms, suggesting a checkpoint defect in NF1-deficient cells.Citation33,Citation34 Thus, understanding checkpoint function in IRA-deficient yeast cells could increase our knowledge of the process of primary tumor formation in NF1 and the role of NF1 deficiency in tumors that arise after radiotherapy.
The aim of this work was to investigate whether loss of the NF1 homologs in yeast altered the DNA damage checkpoint. Using a genetic system to transiently activate the DNA damage checkpoint pathway, we found that a yeast strain lacking the IRA genes was able to initiate and maintain a checkpoint-mediated arrest. However, IRA deficient strains were unable to recover from the arrest, despite dephosphorylation of Rad53. We show that the recovery defect requires checkpoint activation and PKA phosphorylation sites on Cdc20. Our data suggest a model where the DNA damage checkpoint pathway recruits PKA signaling to re-enforce a preanaphase arrest. When PKA regulatory elements are hyperactivated by IRA deletion, cells are unable to restore normal PKA signaling after checkpoint downregulation, leading to a permanent mitotic arrest. Recently, Ras signaling has also been implicated in localization of signaling factors required for mitotic exit.Citation35,Citation36 In IRA deletion strains, activated Ras and PKA could therefore disrupt several layers of mitotic regulation. This work defines a new signaling pathway that can regulate recovery from the DNA damage checkpoint and a second pathway that compromises recovery without causing sustained activation of the DNA damage checkpoint signal.
Results
IRA deletion causes a defect in recovery from DNA damage checkpoint arrest.
To determine whether Ras deregulation causes sensitivity to DNA damage, we generated cdc13-1 strains with deletion of one or both IRA genes. CDC13 encodes a telomere binding protein that maintains telomere structure and recruits telomerase to maintain telomere length. cdc13-1 encodes a temperature sensitive variant that dissociates from telomeres at the nonpermissive temperature. Cdc13 dissociation causes telomeric DNA to be recognized by the DNA damage checkpoint, triggering cell cycle arrest at the metaphase-to-anaphase transition. cdc13-1 strains grew at 25° and 26° but failed to grow at 30°. With deletion of one or both IRA genes, the cdc13-1 strain grew poorly at 25° and failed to grow at 26° (). This result suggested that a strain lacking the IRA genes is sensitive to DNA damage caused by deprotection of the telomeres. In accordance with a published report from a high throughput study,Citation37 we also observed an increase in sensitivity to the DNA alkylating agent methyl methanesulfonate in CDC13 strains with IRA deletion, suggesting this phenotype is also associated with chemically-induced DNA damage (Fig. S1).
One explanation for increased sensitivity to DNA damage is a failure of the DNA damage checkpoint to arrest the yeast cell cycle. A microcolony assay was performed to determine whether an ira1Δ ira2Δ strain was appropriately arresting in response to cdc13-1 inactivation. When incubated at 30° for 8 hours, cdc13-1 cells with an intact DNA damage checkpoint arrest within one cell cycle, forming colonies with 2–4 cells. Checkpoint defective strains continue dividing despite the DNA damage and form colonies of more than four cells. There was no difference in the microcolony size distribution of a cdc13-1 ira1Δ ira2Δ strain and a cdc13-1 IRA1 IRA2 control (). 85% ± 3% of cdc13-1 microcolonies consisted of 4 or fewer cells, compared to 90% ± 7% of cdc13-1 ira1Δ ira2Δ (p = 0.34). This result indicated that the IRA deletion strain did not have a defect in cell cycle arrest following cdc13-1 inactivation.
We next considered that the ira1Δ ira2Δ strain might have a defect in recovery from transient cdc13-1 inactivation. When synchronized cultures were raised to the cdc13-1 non-permissive temperature, then allowed to recover at 21°, viability of the cdc13-1 ira1Δ ira2Δ strain was significantly reduced (). After three hours of cdc13-1 inactivation, 11% ± 4% of ira1Δ ira2Δ cells were able to form colonies, compared to 57% ± 15% of IRA1 IRA2 controls (p < 0.01). The recovery defect is not caused by IRA deletion alone since a CDC13 ira1Δ ira2Δ grows well after 4, 8 and 12 hours of incubation at 32° (Fig. S2). This result indicated that deletion of the IRA genes compromises DNA damage checkpoint recovery.
To further characterize the nature of the checkpoint recovery defect, colonies were inspected microscopically after an 18-hour recovery period. In the cdc13-1 control strain, most cells formed healthy colonies in the recovery period (). With increasing cdc13-1 inactivation time, many control colonies had abnormal morphology but still consisted of more than four cells. In contrast, cdc13-1 ira1Δ ira2Δ cells frequently remained in the 2–4 cell stage during the recovery period. After three hours of cdc13-1 inactivation, 75% of cdc13-1 ira1Δ ira2Δ colonies consisted of 2–4 cells compared to only 1.3% of cdc13-1 IRA1 IRA2. In the ira1Δ ira2Δ strain the fraction of 2–4 cell colonies increased with longer cdc13-1 inactivation and was tightly correlated with the loss of viability (Fig. S3). This result suggested that in the absence of the IRA genes, cells have a significant delay in resuming the cell cycle after transient cdc13-1 inactivation, which ultimately compromises colony forming ability.
Ras deregulation cannot restrain mitosis in checkpoint-deficient cells.
Delayed recovery from cdc13-1-mediated arrest suggested that Ras deregulation could delay mitosis in checkpoint-proficient cells having a DNA damage signal from unstructured telomeres. To determine whether Ras-mediated cell cycle delay required an intact DNA damage checkpoint, we tested whether Ras deregulation could delay the cell cycle in checkpoint defective cells. Microcolony assays were performed with cdc13-1 strains having CHK1 deletion, PDS1 deletion, or carrying a hypomorphic allele of RAD53. Deletion of IRA1 and IRA2 did not improve the checkpoint defect of cdc13-1 chk1Δ, cdc13-1 rad53-21, or cdc13-1 pds1Δ cells (). In fact, IRA deletion slightly worsened the checkpoint defect of a cdc13-1 chk1Δ strain. In the cdc13-1 chk1Δ strain about 20% of colonies had 13 or more cells, compared to more than 60% of colonies in the cdc13-1 chk1Δ ira1Δ ira2Δ strain. This might be caused by acceleration of the G1-S transition or mitotic exit in chk1Δ cells, or alternatively if IRA deletion slows the cell cycle, viability of the cells could increase leading to more cells per microcolony. We also observed a mild but significant increase in the number of microcolonies containing more than 4 cells in cdc13-1 ira1Δ ira2Δ rad53-21 compared to cdc13-1 rad53-21 controls (). In cdc13-1 ira1Δ ira2Δ rad53-21, 84.3% ± 3.2% of colonies had >4 cells, compared to 76.0% ± 1.7% in cdc13-1 rad53-21 (mean ± standard deviation of three assays, p = 0.016 by two-tailed Student's t-test). Therefore, in both chk1Δ and rad53-21 cells, IRA deletion increased the number of cells per microcolony. In both cases this effect could be due to an accelerated cell cycle allowing for more cell divisions during the incubation period, or possibly from a cell cycle delay that increases cell viability and allows for more divisions producing viable cells. We concluded that Ras deregulation caused by IRA deletion is not able to delay cell cycle progression in a checkpoint-deficient background.
ira1Δ ira2Δ cells are proficient in dephosphorylating Rad53.
A number of DNA damage recovery defective yeast mutants have been described. Frequently, such strains fail to recover from DNA damage-induced arrest because they are unable to inactivate the checkpoint kinase Rad53. Surprisingly, we found that the recovery-defective cdc13-1 ira1Δ ira2Δ strain was proficient in Rad53 dephosphorylation, indicating the Rad53 branch of the checkpoint was appropriately turned off in this strain. In both IRA1 IRA2 and ira1Δ ira2Δ strains, Rad53 was dephosphorylated after 60 to 120 minutes of recovery from a four-hour cdc13-1 inactivation in synchronized cells (). There are two Rad53 bands in recovered cells due to a mitosis specific phosphorylation of Rad53, which does not activate its kinase activity.Citation38,Citation39 We confirmed that the T = 180 Rad53 shift was equivalent to that seen in nocodazole-treated cells, which arrest in mitosis without activating Rad53 (). Despite equal kinetics of checkpoint inactivation, cdc13-1 ira1Δ ira2Δ cells failed to progress through mitosis up to three hours after recovery (). While cdc13-1 cells began entering anaphase after 120 minutes of recovery, shortly after Rad53 dephosphorylation, cdc13-1 ira1Δ ira2Δ cells maintained the preanaphase arrest for up to 180 minutes. The delay in mitotic progression despite Rad53 dephosphorylation was confirmed in triplicate assays (Fig. S4). We concluded that persistent checkpoint signaling is unlikely to be responsible for the recovery defect in cdc13-1 ira1Δ ira2Δ cells. Although we did not directly examine the phosphorylation state of Chk1 in these strains, we consider it unlikely that persistent Chk1 activation is responsible for the cell cycle delay (see Discussion).
IRA deletion causes permanent cell cycle arrest in cells recovering from DNA damage.
We observed an increase in the number of 2–4 cell colonies in the cdc13-1 ira1Δ ira2Δ strain after 18 hours of recovery from cdc13-1 inactivation (); however, this result does not establish whether the arrested cells at 18 hours eventually resume cell division. In order to examine the permanency of the checkpoint recovery defect, we adapted an approach to monitor individual cells over several days of recovery from a DNA damage mediated arrest.Citation40 Individual G1 cells were micromanipulated onto 32° YPD agar, maintained at 32° for three hours, then returned to 21°. Colonies were inspected microscopically after 6, 24, 48 and 72 hours of recovery and classified as visible to the naked eye (Recovered), microscopically visible consisting of 3 or more cells (Microscopic), or as a single large-budded cell (2-cell). The kinetics of checkpoint recovery differed between cdc13-1 and cdc13-1 ira1Δ ira2Δ strains (). More than 90% cdc13-1 cells re-budded within 6 hours of recovery and the vast majority of these cells went on to form visible colonies within 72 hours (). In contrast, fewer than half of cdc13-1 ira1Δ ira2Δ cells re-budded within 6 hours. While some of these late-recovering cells eventually re-budded, about half (25% of the total population) never re-budded and were still 2-cell colonies after 72 hours of recovery (). This result suggested that most ira1Δ ira2Δ cells have a delay in mitotic exit, with a subset completely failing to re-enter the cell cycle after cdc13-1 arrest. Permanent 2-cell colonies were extremely rare in cdc13-1 cells, with only one such event observed in over 150 cells examined across three separate assays. The permanent 2-cell arrest required cdc13-1. When treated similarly, >95% of CDC13 ira1Δ ira2Δ cells formed visible colonies after 72 hours of recovery (data not shown). We used the frequency of permanent 2-cell arrest in subsequent assays to characterize the signaling components required for the Ras-mediated checkpoint recovery defect. This criteria is more stringent than the scoring approach used in , which grouped colonies having 2–4 cells together and did not account for late-recovering cells. This result indicated that the cell cycle arrest of ira1Δ ira2Δ cells after cdc13-1 inactivation is a permanent state, with cells remaining in the 2-cell stage for several days after the DNA damage signal is deactivated.
Permanent cell cycle arrest requires Tpk2, Chk1 and PKA phosphorylation of Cdc20.
Deletion of IRA1 and IRA2 leads to increased Ras2 activation, which increases intracellular cAMP and promotes signaling through the cAMP-dependent protein kinase (PKA) pathway. PKA has been shown to participate in the DNA damage checkpoint. We therefore sought to determine whether the recovery defect in the ira1Δ ira2Δ strain was due to deregulation of PKA. Strains were treated as described for the single-cell recovery assay and colonies were examined after 72 hours of recovery. In this set of assays, 48.1% ± 9.6% of cdc13-1 ira1Δ ira2Δ cells were permanently arrested, compared to less than 1% of control cdc13-1 cells (). With deletion of the PKA catalytic subunit-encoding TPK2 gene, the 2-cell fraction dropped to 14.8% ± 3.0% (p < 0.05 compared to cdc13-1 ira1Δ ira2Δ) indicating that Tpk2 is required for the full recovery defect in the IRA deletion strain.
We next asked whether the recovery defect in ira1Δ ira2Δ cells was checkpoint-dependent. We found that deletion of CHK1 significantly rescued the 2-cell arrest in cdc13-1 ira1Δ ira2Δ cells (). While 24.5% ± 6.6% of cdc13-1 ira1Δ ira2Δ cells were permanently arrested, only 4.8% ± 0.7% of cdc13-1 chk1 ira1Δ ira2Δ cells had the phenotype (p < 0.05). This result supports a model where a Chk1-dependent cell cycle arrest is required for the ira1Δ ira2Δ recovery defect. This result also ruled out the possibility that permanent 2-cell arrest was caused by other conditions such as increased temperature or α-factor recovery used during the experiment.
During a DNA damage-induced arrest, PKA can re-enforce a cell cycle delay by phosphorylating Cdc20 and preventing ubiquitin-mediated degradation of the anaphase inhibitor Pds1 and the mitotic cyclin Clb2 by the anaphase promoting complex (APC).Citation10 We reasoned that IRA deletion might compromise DNA damage checkpoint recovery by deregulating PKA, leading to constitutive APCCdc20 inhibition. This model predicted that preventing Cdc20 phosphorylation by PKA would be sufficient to rescue the recovery defect. To test this model we introduced plasmids containing wild-type CDC20 or an allele with mutation in two PKA phosphorylation sites, S52 and S88, into cdc13-1 IRA1 IRA2 or cdc13-1 ira1Δ ira2Δ strains.Citation10 The mutant CDC20 allele significantly rescued the 2-cell arrest in the ira1Δ ira2Δ strain, lowering the 2-cell population from 40.1% ± 6.2% to 6.2% ± 2.5% (p < 0.01, ). Thus, the recovery defect in an ira1Δ ira2Δ strain requires PKA phosphorylation of sites on Cdc20.
In summary, we found that a yeast strain lacking the Ras GTPase activating proteins Ira1 and Ira2 is defective in DNA damage checkpoint recovery. The defect requires Chk1-mediated cell cycle arrest, PKA catalytic activity and PKA phosphorylation of Cdc20. Our data suggest that PKA can inappropriately restrain mitosis by re-enforcing cell cycle arrest in a checkpoint- and Pds1-dependent manner after a transient DNA damage stimulus. When cells activate the DNA damage checkpoint in the setting of deregulated Ras, recovery from the checkpoint can be delayed or completely prevented.
Discussion
Nutrient-sensing pathways play a role in DNA damage checkpoint recovery.
Eukaryotic cells respond to DNA damage by activating checkpoint pathways that allow cells to cope with genotoxic stress. Recently, signaling pathways that respond to nutrients have been implicated in the DNA damage checkpoint. In budding yeast the Ras-cAMP-PKA pathway responds to glucose and generally promotes cell growth and division.Citation41 PKA can support a preanaphase arrest in checkpoint deficient cells experiencing a DNA damage signal. In the work presented here we tested whether increased Ras signaling could modify the DNA damage response in cells with an intact checkpoint. We have shown that (1) cells with high Ras activity are defective in checkpoint recovery, (2) the recovery defect requires an intact checkpoint and PKA signaling to Cdc20 and (3) the recovery defect is different from most other recovery-defective strains because the checkpoint signaling pathway is appropriately down-regulated in Ras de-regulated cells.
Signaling requirements for a Ras-mediated recovery defect.
When one or both IRA genes are deleted, two downstream pathways are activated: the PKA pathway and a MAPK pathway promoting cell morphology changes. Our data suggest that the PKA pathway is the branch required for a recovery defect, since deletion of the PKA catalytic subunit TPK2 partially rescued the recovery defect. Incomplete rescue may be due to functional overlap between the three PKA catalytic subunits (TPK-1, -2 and -3) encoded in yeast. Both TPK1 and TPK2 can participate in checkpoint-mediated preanaphase arrest,Citation10 and all three subunits can regulate mitosis under different nutrient conditions.Citation42 We consider it likely that Tpk1 is able to prevent recovery, although less efficiently, in the absence of TPK2.
PKA supports preanaphase arrest in DNA-damaged cells lacking CHK1.Citation10 We found that increased Ras-PKA signaling prevented DNA damage checkpoint recovery, resulting in a loss of viability with cdc13-1 inactivation. The Ras deregulated strain characteristically remained arrested in the 2-cell state after the DNA damage stimulus was removed. In cells lacking CHK1, the population of permanently arrested cells was significantly diminished, indicating that cells unable to fully activate the checkpoint are less susceptible to the recovery defect. We propose that the recovery defect of Ras deregulated cells requires both PKA catalytic activity and robust checkpoint activation.
We predicted that the anaphase inhibitor Pds1 would be required for the recovery defect in cdc13-1 ira1Δ ira2Δ. The single-cell recovery approach was not suitable in pds1Δ strains because the cells re-budded during the cdc13-1 inactivation period. This result suggested that IRA deletion was not delaying mitosis in the absence of Pds1. Consistent with this observation, IRA deletion did not rescue the checkpoint defect of a cdc13-1 pds1Δ strain in a microcolony assay, further demonstrating that the cell cycle delay in Ras de-regulated cells requires Pds1. This is in accordance with previous work showing that high cAMP inhibits mitosis in a Pds1-dependent manner during nutrient-limiting conditions.Citation43
PKA regulates mitosis by modulating the anaphase promoting complex specificity factor Cdc20. PKA phosphorylation of Cdc20 at two serine residues is required for PKA to restrain mitosis in a chk1Δ cell. A mutant CDC20 allele defective for PKA-mediate phosphorylation significantly rescued the recovery defect in cdc13-1 ira1Δ ira2Δ cells, supporting a model where PKA is recruited by the fully-activated DNA damage checkpoint to phosphorylate Cdc20 and prevent mitotic progression by preventing Pds1 degradation.
Taken together, our data suggest that Ras deregulation acts through PKA to inhibit mitosis in recovering cells by preventing Cdc20-mediated degradation of Pds1. According to our model, in a normal cell with an intact checkpoint, PKA signaling is rewired to target mitotic substrates. When the checkpoint is deactivated, the PKA regulatory mechanisms, including Ira1 and Ira2, act to restore basal cAMP-PKA status to allow mitotic progression. In the absence of IRA1 and IRA2, PKA signaling cannot be reset after the checkpoint-dependent rewiring, leading to permanent cell cycle arrest. We do not know whether high cAMP levels are maintained during the arrest, or if the PKA catalytic activity becomes uncoupled from cAMP availability. However, since IRA deletion strains are known to have prolonged cAMP elevation after glucose stimulation of starved cells,Citation14,Citation15 the former situation seems more likely.
Rad53 is dephosphorylated in ira1Δ ira2Δ cells.
To determine if the DNA damage checkpoint is being appropriately turned off in IRA deletion cells, we examined Rad53 phosphorylation status, a marker of checkpoint activation. We found that Rad53 was dephosphorylated with wild-type kinetics in ira1Δ ira2Δ cells, although the cells did not progress through mitosis in the time frame examined. This result does not directly address a role for Chk1 in maintaining the checkpoint arrest in these strains; however, we consider this unlikely. We have shown that signaling through Chk1 alone is not sufficient to maintain DNA damage induced preanaphase delay.Citation7 In addition the Haber group showed that degradation of Rad53 allowed cell division after a double stranded break in an adaptation deficient strain, suggesting that Rad53 inactivation was sufficient for resuming the cell cycle during adaptation.Citation24 In the present work, a cdc13-1 ira1Δ ira2Δ strain containing a Cdc20 allele with mutations in two PKA phosphorylation sites was found to be recovery-proficient. The mutant form of Cdc20 cannot override the DNA damage arrest in a cell with an intact Chk1 pathway.Citation10 Therefore, the ability of Cdc20 mutant cells to recover from cdc13-1 arrest suggests that the Chk1 branch of the checkpoint pathway is inactivated in these cells.
Possible mechanisms for PKA-mediated cell cycle arrest.
It is tempting to speculate that physical relocalization of the PKA holoenzyme is responsible for the recovery defect in IRA deletion cells. PKA kinase activity is negatively regulated by the regulatory subunit, encoded by BCY1. Phosphorylation of the PKA regulatory subunit is known to influence its subcellular localization during heat stress and nutrient deprivation and recent work in our laboratory indicates that R subunit phosphorylation is required for PKA to delay mitotic progression in chk1Δ cells (Searle et al. submitted). How might this model explain the recovery defect? The PKA holoenzyme might be recruited to nuclear substrates, with constitutive Ras activation generating high cAMP, leading to chronic activation of the catalytic subunits. Conversely, relocalization of the R subunits to the cytoplasm during DNA damage could physically separate the C and R subunits, allowing high PKA activity in the nucleus. It is also possible that the holoenzyme localizes to cytoplasmic cell cycle regulatory complexes located at the bud neck and spindle pole body. These models are not mutually exclusive. Future work will define what aspects of PKA regulation contribute to the recovery defect in IRA deletion cells. We recently defined these mechanisms for PKA's role in re-enforcing the checkpoint (Searle et al. submitted). It will be interesting to learn whether the recovery defect represents a persistent over-activation of the same mechanism, or if different processes are involved in the recovery defect. For example, the aurora kinase Ipl1 and PKA have overlapping substrate specificities. Ipl1 can phosphorylate kinetochore substrates to regulate kinetochore-microtubule attachment.Citation44,Citation45 PKA can phosphorylate targets at the kinetochore, which might modify spindle attachment dynamics.Citation46 When PKA is recruited in the DNA damage checkpoint, there may be inappropriate phosphorylation of kinetochore substrates, which would inhibit microtubule attachment and delay mitosis through activation of the spindle checkpoint. We are currently investigating whether the spindle checkpoint has a role in the PKA-mediated recovery defect.
Potential roles for PKA in neurofibromatosis type 1 associated tumors.
The IRA genes are homologs of the human NF1 tumor suppressor, a Ras-GAP that is mutated in the tumor predisposition syndrome neurofibromatosis type 1 (NF1). The disease is highly pleiotrophic, but one severe consequence of NF1 is a predisposition toward malignant peripheral nerve sheath tumors (MPNST) arising from Schwann cell progenitors. Schwann cells proliferate in response to cAMP and NF1-deficient Schwann cells have high cAMP levels, although unlike yeast this increase is not mediated by Ras. If the role for PKA in the DNA damage response is conserved in mammalian cells, the Schwann cell component of a nerve sheath tumor might respond differently to DNA damage. Since MPNSTs respond poorly to chemotherapy, it is possible that malignant cells have adapted to checkpoint deregulation and consequently are less susceptible to DNA damage. In summary, we identified a new role for Ras and PKA in regulating the DNA damage response in budding yeast. This signaling pathway might contribute to tumor progression in individuals with neurofibromatosis type 1.
Materials and Methods
Strain growth and media.
Yeast strains were grown in standard yeast extract/peptone/dextrose (YPD) media. α-factor synchronization was performed in YPD pH 3.9. When plasmid selection was required, strains were grown in synthetic complete media lacking leucine (C-Leu). Unless noted, strains were grown at 21°.
Strain and plasmid generation.
Strains and plasmids used in this study are shown in . All strains are derived from the Y300 background.Citation47 ira1Δ::URA3 and ira2Δ::URA3 in Y300 were generated by one-step gene deletion using a knockout cassette generated by PCR from the pRS416 plasmid. Primers were designed with homology to the 5′ and 3′ UTR of IRA1 or IRA2 and homology to pRS416, such that the PCR product contained the URA3 marker and ∼40 bp of homology to the IRA flanking regions (sequences are available upon request). The knockout cassette was transformed by standard lithium acetate/PEG/heat shock, except for cdc13-1 strains where the heat shock was omitted and cells were incubated in Li-PEG for 18 hours before plating. IRA deletion strains were back-crossed to the Y300 background to generate mating type α progeny, which were then crossed to Y818 or Y831 and sporulated to obtain cdc13-1 ira1Δ ira2Δ chk1Δ and cdc13-1 ira1Δ ira2Δ rad53-21. YMW544 and YMW545 were obtained by crossing YMW199 to a pds1Δ::LEU2 α Y300 strain. The tpk2Δ::KAN allele was back-crossed to the Y300 background from the Open Biosystems yeast deletion strain, then mated to YMW142 and YMW199 to obtain YMW257 and YMW512, respectively. Plasmids pMW034 and pMW036 were constructed by SmaI-SpeI digest of pJS3 (HA-CDC20-WT) or pJS6 (HA-CDC20-S52A-S88A) and cloning of the fragment into SmaI-SpeI digested pRS415. Correct insertions were confirmed by restriction digest with HinDIII. Plasmids were transformed into YMW446, with Leu+ transformants sporulated and dissected to obtain YMW534-YMW537.
Drop assays.
Log phase cultures were diluted to OD(600) 0.08 (∼106 cells per mL) and 10-fold serial dilutions were made. 5 µL drops were placed on room temperature YPD agar. Plates were incubated at 25, 26 or 30° for three days before photographing.
Microcolony assays.
Log phase cultures were grown overnight at 21°. Cultures were diluted to 50,000 cells per mL, sonicated and 100 µL was spread to YPD agar plates prewarmed to 30°. Plates were incubated at 30° for 8 hours. The number of cells in 100 colonies was counted.
cdc13-1 recovery assay.
Strains were arrested with α-factor in YPD pH 3.9 for 4.5 hours. Cultures were raised to 32° for 75 minutes to inactivate cdc13-1, then cells were released from the α-factor block by three washes with 32° YPD pH 6.2 media. At the indicated times after α-factor release, samples were extracted, sonicated, counted with a hemocytometer, serially diluted and plated to room temperature YPD agar. Viability was calculated as colony-forming units per mL divided by total cells per mL, normalized to the viability of α-factor samples.
Rad53 dephosphorylation and visualization of nuclei.
Log phase cultures were synchronized with α -factor in YPD pH 3.9 for 4.5 hours at 21°, then raised to 32° for 75 minutes before washing out α-factor with 32° YPD pH 6.2. Cultures were held at 32° for 4 hours, then returned to 21°. Protein samples were isolated through TCA precipitation. Samples were fractionated on 10% acrylamide 0.067% bis-acrylamide gels, transferred to nitrocellulose membranes and probed for Rad53 by western analysis (Santa Cruz Biotechnology, Sata Cruz CA clone yC-19 1:500 in 5% BSA TBS-0.05% Tween, secondary Jackson donkey-anti-goat HRP 1:20,000 in 5% milk TBS-Tween). In parallel, cells were fixed and permeablized in 70% ethanol, rehydrated with PBS and stained with 1 µg/mL 4′6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma, St. Louis, MO). Cells were mounted on 0.1% poly(L)-lyseine coated slides for microscopic visualization.
Single-cell recovery assays.
Log phase 21° YPD cultures were diluted to OD = 0.15, then arrested with α-factor for 4.5 hours at 21° in YPD pH 3.9. Cultures were raised to 32° for 75 minutes to inactivate Cdc13, then a sample was sonicated and dropped onto 32° YPD agar. Fifty-five cells with a Shmoo were micro-manipulated onto the agar, and the plates were returned to 32° for three hours before removing to 21° (T = 0 hours, start of the recovery period). Colonies that had not budded during the cdc13-1 arrest period were presumed killed by the micromanipulation and were excluded from analysis (generally ∼10% of the cells). Colony morphologies were recorded at 6/18/48/72 hours for kinetics, or at 72 hours for terminal phenotype analysis.
Statistical analysis.
Values shown are mean ± standard deviation or standard error of at least three independent assays. Statistical tests were performed in Microsoft Excel with a 2-tailed Student's t-test assuming equal variance.
Abbreviations
| PKA | = | cAMP dependent protein kinase |
| APC | = | anaphase promoting complex |
| NF1 | = | neurofibromatosis type 1 |
| MPNST | = | malignant peripheral nerve sheath tumor |
Figures and Tables
Figure 1 IRA deletion strains are sensitive to cdc13-1-induced arrest despite an intact DNA damage checkpoint. (A) 10-fold serial dilutions were spotted to YPD agar and incubated at the indicated temperature for three days. (B) Log phase cultures were sonicated, spread onto 30° YPD agar and incubated at 30° for 8 hours. 100 colonies were scored for each strain. Values are mean ± standard deviation of three independent assays. (C) Cultures were arrested with α-factor at 21°, then raised to 32° to inactivate cdc13-1, then released from the α-factor block at 32°. Samples were sonicated, counted, serially diluted, and plated to 21° YPD agar. Viability is calculated as (CFU/mL)/(Total cells per mL) normalized to α-factor samples. Values are mean ± standard deviation of three assays. **p < 0.01, ***p < 0.001 by student's t-test. (D) YPD plates from (C) were inspected 18 hours after returning to 21°. Between 50 and 60 colonies were scored for each strain. Values are mean ± standard deviation of three assays.
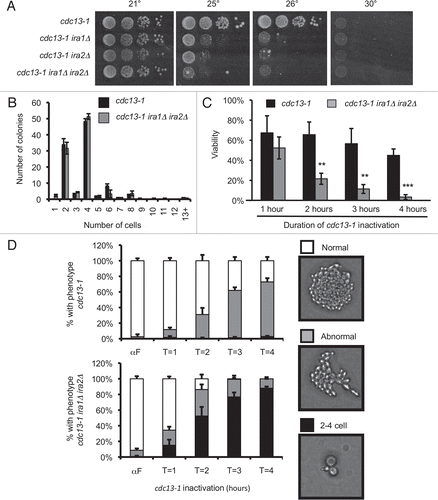
Figure 2 IRA deletion cannot restrain mitosis in the absence of an intact checkpoint. Cultures were treated as in . Values are mean ± standard deviation of three independent assays. Graphs represent microcolony assays for (A) chk1Δ, (B) rad53-21 and (C) pds1Δ backgrounds.
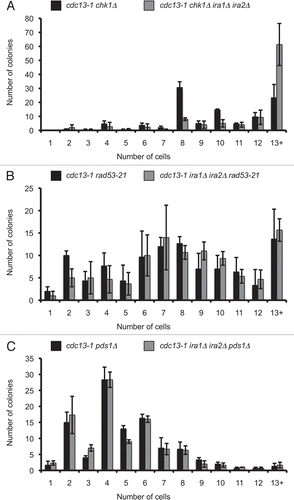
Figure 3 IRA deletion strains dephosphorylate Rad53 under conditions that delay checkpoint recovery. Cultures were synchronized with α-factor in YPD pH 3.9, raised to 32° for 75 minutes, and α-factor was removed by washing cells with 32° YPD pH 6.2. Cultures were held at 32° for four hours, then returned to 21°. Protein samples were harvested by TCA precipitation and analyzed by western analysis for Rad53 phosphorylation (A) and cells were fixed and analyzed for nuclear division after 0–180 minutes of recovery (C). In (B), samples were run adjacent to extracts from cultures treated with 10 µg/mL nocodazole for 4 hours at 21°. >90% of cells were large-budded at the time of harvest.
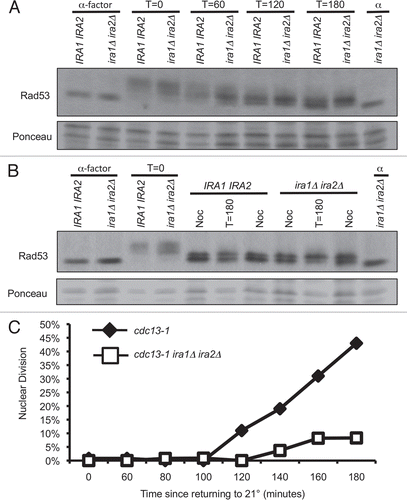
Figure 4 cdc13-1 ira1Δ ira2Δ has delayed recovery kinetics. α-factor arrested cultures were raised to 32° and released from the G1 arrest by micromanipulation onto 32° YPD agar. After three hours at 32°, plates were returned to 21° (T = 0 hours recovery). Colonies were scored at the indicated times and classified as visible to the naked eye (Recovered), microscopically visible with more than 2 cells (Microscopic) or as single large-budded cells (2-cell). Values are mean ± standard error of three assays. (A) cdc13-1, (B) cdc13-1 ira1Δ ira2Δ.
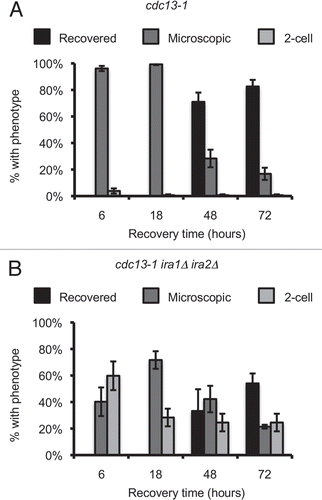
Figure 5 Single-cell recovery assays reveal signaling elements required for the ira1Δ ira2Δ recovery defect. (A and B) Strains were treated as in . Colonies were scored after 72 hours of recovery. (C) Strains were treated as in except cultures were in synthetic complete media lacking leucine (C-Leu) to maintain CDC20 plasmids. Values are mean ± standard error of at least three assays.
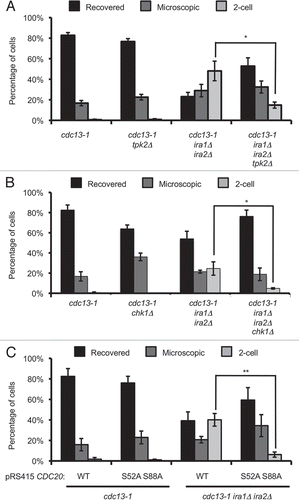
Table 1 Strains and constructs used in this study
Additional material
Download Zip (2.7 MB)Acknowledgements
We thank members of the Sanchez Laboratory for their insightful comments on this work and Deborah Hogan for comments on the manuscript. This work was funded by NIH/NCI R01CA84463 and NIH/NINDS R21NS060940 to YS (http://www.cancer.gov/, http://www.ninds.nih.gov/), NIH training grant T32-CA009658-18 (http://www.nih.gov/) and by a Children's Tumor Foundation Young Investigator Award to M.D.W. (http://www.ctf.org/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. M.D.W. is an Albert J. Ryan fellow. The authors declare no competing financial interests.
References
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science 1996; 274:1664 - 1672
- Hartwell LH, Kastan MB. Cell cycle control and cancer. Science 1994; 266:1821 - 1828
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433 - 439
- Chen Y, Sanchez Y. Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA Repair 2004; 3:1025 - 1032
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997; 277:1497 - 1501
- Agarwal R, Tang Z, Yu H, Cohen-Fix O. Two distinct pathways for inhibiting pds1 ubiquitination in response to DNA damage. J Biol Chem 2003; 278:45027 - 45033
- Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, et al. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 1999; 286:1166 - 1171
- Wang H, Liu D, Wang Y, Qin J, Elledge SJ. Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev 2001; 15:1361 - 1372
- Hu F, Wang Y, Liu D, Li Y, Qin J, Elledge SJ. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 2001; 107:655 - 665
- Searle JS, Schollaert KL, Wilkins BJ, Sanchez Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat Cell Biol 2004; 6:138 - 145
- Park JI, Grant CM, Dawes IW. The high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae is the major determinant of cAMP levels in stationary phase: involvement of different branches of the Ras-cyclic AMP pathway in stress responses. Biochem Biophys Res Commun 2005; 327:311 - 319
- Kataoka T, Powers S, Cameron S, Fasano O, Goldfarb M, Broach J, et al. Functional homology of mammalian and yeast RAS genes. Cell 1985; 40:19 - 26
- Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Broek D, et al. In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell 1985; 40:27 - 36
- Tanaka K, Matsumoto K, Toh EA. IRA1, an inhibitory regulator of the RAS-cyclic AMP pathway in Saccharomyces cerevisiae. Mol Cell Biol 1989; 9:757 - 768
- Tanaka K, Nakafuku M, Tamanoi F, Kaziro Y, Matsumoto K, Toh-e A. IRA2, a second gene of Saccharomyces cerevisiae that encodes a protein with a domain homologous to mammalian ras GTPase-activating protein. Mol Cell Biol 1990; 10:4303 - 4313
- Tanaka K, Nakafuku M, Satoh T, Marshall MS, Gibbs JB, Matsumoto K, et al. S. cerevisiae genes IRA1 and IRA2 encode proteins that may be functionally equivalent to mammalian ras GTPase activating protein. Cell 1990; 60:803 - 807
- Harashima T, Anderson S, Yates JR 3rd, Heitman J. The kelch proteins Gpb1 and Gpb2 inhibit Ras activity via association with the yeast RasGAP neurofibromin homologs Ira1 and Ira2. Mol Cell 2006; 22:819 - 830
- Clemenson C, Marsolier-Kergoat MC. DNA damage checkpoint inactivation: adaptation and recovery. DNA Repair 2009; 8:1101 - 1109
- Bazzi M, Mantiero D, Trovesi C, Lucchini G, Longhese MP. Dephosphorylation of gamma H2A by Glc7/protein phosphatase 1 promotes recovery from inhibition of DNA replication. Mol Cell Biol 30:131 - 145
- Guillemain G, Ma E, Mauger S, Miron S, Thai R, Guerois R, et al. Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol Cell Biol 2007; 27:3378 - 3389
- Leroy C, Lee SE, Vaze MB, Ochsenbien F, Guerois R, Haber JE, et al. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol cell 2003; 11:827 - 835
- Sandell LL, Zakian VA. Loss of a yeast telomere: arrest, recovery and chromosome loss. Cell 1993; 75:729 - 739
- Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997; 90:1097 - 1106
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/Marrest. Mol cell 2001; 7:293 - 300
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, et al. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol cell 2002; 10:373 - 385
- Schwartz DC, Felberbaum R, Hochstrasser M. The Ulp2 SUMO protease is required for cell division following termination of the DNA damage checkpoint. Mol Cell Biol 2007; 27:6948 - 6961
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990; 63:851 - 859
- Buchberg AM, Cleveland LS, Jenkins NA, Copeland NG. Sequence homology shared by neurofibromatosis type-1 gene and IRA-1 and IRA-2 negative regulators of the RAS cyclic AMP pathway. Nature 1990; 347:291 - 294
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1?. Glia 2008; 56:1590 - 1605
- Kim HA, Ratner N, Roberts TM, Stiles CD. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J Neurosci 2001; 21:1110 - 1116
- Sheela S, Riccardi VM, Ratner N. Angiogenic and invasive properties of neurofibroma Schwann cells. The J Cell Biol 1990; 111:645 - 653
- Xu Y, Chiamvimonvat N, Vazquez AE, Akunuru S, Ratner N, Yamoah EN. Gene-targeted deletion of neurofibromin enhances the expression of a transient outward K+ current in Schwann cells: a protein kinase A-mediated mechanism. J Neurosci 2002; 22:9194 - 9202
- Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet 2006; 43:289 - 294
- Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol 2006; 24:2570 - 2575
- Seshan A, Amon A. Ras and the Rho effector Cla4 collaborate to target and anchor Lte1 at the bud cortex. Cell Cycle 2005; 4:940 - 946
- Yoshida S, Ichihashi R, Toh-e A. Ras recruits mitotic exit regulator Lte1 to the bud cortex in budding yeast. J Cell Biol 2003; 161:889 - 897
- Hanway D, Chin JK, Xia G, Oshiro G, Winzeler EA, Romesberg FE. Previously uncharacterized genes in the UV- and MMS-induced DNA damage response in yeast. Proc Natl Acad Sci U S A 2002; 99:10605 - 10610
- Schleker T, Shimada K, Sack R, Pike BL, Gasser SM. Cell cycle-dependent phosphorylation of Rad53 kinase by Cdc5 and Cdc28 modulates checkpoint adaptation. Cell Cycle 9:350 - 363
- Clemenson C, Marsolier-Kergoat MC. The spindle assembly checkpoint regulates the phosphorylation state of a subset of DNA checkpoint proteins in Saccharomyces cerevisiae. Mol Cell Biol 2006; 26:9149 - 9161
- Dotiwala F, Haase J, Arbel-Eden A, Bloom K, Haber JE. The yeast DNA damage checkpoint proteins control a cytoplasmic response to DNA damage. Proc Natl Acad Sci USA 2007; 104:11358 - 11363
- Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces responds to nutrients. Annu Rev Genet 2008; 42:27 - 81
- Bolte M, Dieckhoff P, Krause C, Braus GH, Irniger S. Synergistic inhibition of APC/C by glucose and activated Ras proteins can be mediated by each of the Tpk1-3 proteins in Saccharomyces cerevisiae. Microbiology 2003; 149:1205 - 1216
- Anghileri P, Branduardi P, Sternieri F, Monti P, Visintin R, Bevilacqua A, et al. Chromosome separation and exit from mitosis in budding yeast: dependence on growth revealed by cAMP-mediated inhibition. Exp Cell Res 1999; 250:510 - 523
- Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW. The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev 1999; 13:532 - 544
- Pinsky BA, Kung C, Shokat KM, Biggins S. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol 2006; 8:78 - 83
- Li JM, Li Y, Elledge SJ. Genetic analysis of the kinetochore DASH complex reveals an antagonistic relationship with the ras/protein kinase A pathway and a novel subunit required for Ask1 association. Mol Cell Biol 2005; 25:767 - 778
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev 1994; 8:2401 - 2415