Loss of heterozygosity (LOH) is one of the most important genetic mechanisms in tumorigenesis and leads to the inactivation of “so called” tumor suppressor genes that regulate growth and differentiation. In a recent study,Citation1 we discovered in a new mouse tumor model that genes that are essential for cell viability (cell essential genes) can profoundly modify the genetic mechanism of tumorigenesis by impacting LOH at genetically linked tumor suppressor loci. Specifically, we observed that a missense mutation in Rpa1, which is incompatible with cell survival in homozygous mutant cells, prevents LOH at the nearby Trp53 gene locus when it is linked to a mutant Trp53 allele (cis configuration). As a result, the cis-linkage caused selection against Trp53-dependent tumorigenesis by LOH. In contrast to the protective effect that was mediated in cis, when the Rpa1 mutation was located on the opposite chromosome (trans configuration), it enhanced LOH and tumorigenesis. This enhancement was due to the increased genomic instability that is caused by the Rpa1 dominant mutation in the heterozygous state. Our study suggests that these results represent a novel intrinsic genetic mechanism of tumor suppression. Therefore, we propose that a similar mechanism exists in humans, and that specific genetic linkages between functional variants in cell essential genes and mutations in tumor suppressor genes may alter cancer phenotypes based on their allelic configuration. Furthermore, this mechanism may be relevant in other diseases that arise due to LOH.
To date, more than 350 of the 22,000 protein-coding genes in the human genome have been shown to carry recurrent somatic mutations in cancer. In mice, more than 2,000 genes are known to potentially contribute to cancer development.Citation4 More than 1 million single nucleotide polymorphisms (SNPs) have been identified across the human genome, and recent studies have shown that individual human genomes contain a significant number of copy number variants (CNVs) that include deletions and duplications of tens to thousands of kilobases.Citation2,Citation10,Citation11 There is also growing interest in large scale sequencing and DNA copy number analysis of human cancer genomes.Citation2-Citation9 These efforts, which include the Cancer Genome Atlas (cancergenome.nih.gov), have uncovered a large number of genetic variants and CNVs throughout cancer genomes. Importantly, this information can now be used in genome-wide association studies (GWAS) to identify associations with common diseases such as cancer, and some of the pilot projects have already begun to provide exciting new insights into cancer genetics.Citation2-Citation4,Citation11 However, there is very little known about how linkage between cell essential genes and tumor suppressor genes impacts cancer phenotypes. Furthermore, the allelic configurations of disease relevant genes, such as mutated tumor suppressor genes and potential loss of function variants in linked cell essential genes, as well as variants that reduce cell proliferation rates, have not been previously considered in the study of LOH. Our dataCitation1 suggest that in addition to association analysis between SNPs/CNVs and disease phenotypes, such as cancer susceptibility, it will be important to consider the status of essential genes linked to tumor suppressor genes.
Although it is currently unknown how frequent functional SNPs/CNVs occur in cell essential genes, it should not be uncommon given the high prevalence of SNPs/CNVs present throughout the human genome. Since LOH may involve whole chromosomes through the correction of a trisomy that occurs from mitotic non-disjunction or chromosome segments through somatic recombination, lethal mutations in cell essential genes need only be located on the same chromosome to exert a protective effect against a mutation acquired in a tumor suppressor. Therefore, a genome-wide analysis of SNPs/CNVs of cell essential genes and their linkages with tumor suppressor genes may shed new light on an important genetic mechanism in human cancer. A schematic diagram illustrating possible associations between cell essential genes and tumor suppressor genes is depicted in . Importantly, this mechanism may be relevant in both familial cancer syndromes, where affected individuals carry a tumor suppressor mutation in every cell, as well as in sporadic cancers, where mutations are somatically acquired, both of which require a second hit that is often the result of LOH. Finally, the study of phenotypic variability between individuals that carry the same familial mutation will help to establish whether this mechanism of cancer attenuation due to cis-linkage between mutations in cell essential genes and tumor suppressor genes is relevant in human disease.
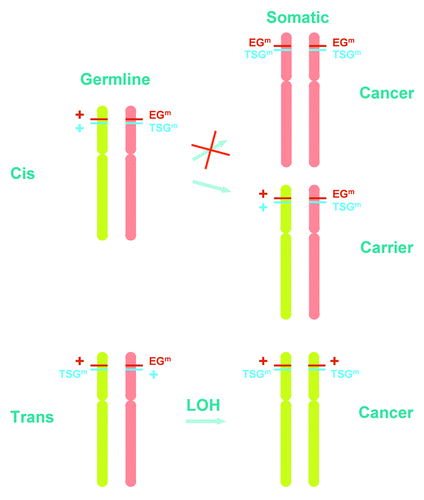
Figure 1. Proposed model for the effect of genetic variants and linkage in disease phenotype. Locations of essential gene (EG) and tumor suppressor gene (TSG) on the chromosomes are representative. The actual linked mapping positions of EG and TSG can be distributed elsewhere along the chromosomes. EGm, mutant cell essential gene; TSGm, mutant tumor suppressor gene. “+” represents wild type EG (red) or wild type TSG (blue). Chromosomes: Green denotes paternal (or maternal) origin and red denotes maternal (or paternal) origin of the chromosome. Cis, EGm and TSGm are on the same chromosome. Trans, EGm and TSGm are on opposite chromosomes. This simplified model assumes whole chromosome loss and gain. However, in reality, the chromosomal changes may be segmental involving fragments of the chromosome. In this case, the EG and TSG segregate within the chromosomal fragment by linkage disequilibrium. This model also assumes LOH is required for tumor formation. However, some mutant tumor suppressor genes may drive tumorigenesis via haploinsufficiency, which is not depicted in the diagram.
References
- Wang Y, Zhang W, Edelmann L, Kolodner RD, Kucherlapati R, Edelmann W. Cis lethal genetic interactions attenuate and alter p53 tumorigenesis. Proc Natl Acad Sci U S A 2010; 107:5511 - 5; http://dx.doi.org/10.1073/pnas.1001223107; PMID: 20212136
- Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science 2008; 322:881 - 8; http://dx.doi.org/10.1126/science.1156409; PMID: 18988837
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008; 9:356 - 69; http://dx.doi.org/10.1038/nrg2344; PMID: 18398418
- Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009; 458:719 - 24; http://dx.doi.org/10.1038/nature07943; PMID: 19360079
- Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009; 462:1005 - 10; http://dx.doi.org/10.1038/nature08645; PMID: 20033038
- Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature 2009; 459:987 - 91; http://dx.doi.org/10.1038/nature08035; PMID: 19536264
- Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al, DIAGRAM Consortium. Parental origin of sequence variants associated with complex diseases. Nature 2009; 462:868 - 74; http://dx.doi.org/10.1038/nature08625; PMID: 20016592
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010; 463:191 - 6; http://dx.doi.org/10.1038/nature08658; PMID: 20016485
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463:899 - 905; http://dx.doi.org/10.1038/nature08822; PMID: 20164920
- Moore JH, Asselbergs FW, Williams SM. Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010; 26:445 - 55; http://dx.doi.org/10.1093/bioinformatics/btp713; PMID: 20053841
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med 2010; 61:437 - 55; http://dx.doi.org/10.1146/annurev-med-100708-204735; PMID: 20059347