Abstract
Histone H2AX phosphorylation on a C-terminal serine residue to form "γ-H2AX" is a critical early event in the chromatin response to chromosomal DNA double strand breaks in eukaryotes. In mammalian cells, γ-H2AX is formed when H2AX is phosphorylated on serine 139 by ATM or by other DNA damage response kinases. H2AX prevents genomic instability and tumorigenesis, and supports class-switch recombination at immunoglobulin heavy chain loci in mammals. We showed previously that H2AX controls double strand break repair by homologous recombination (HR) between sister chromatids. The HR functions of H2AX are mediated by interaction of γ-H2AX with the chromatin-associated adaptor protein MDC1. H2AX is potentially subject to additional post-translational modifications associated with the DNA damage response and with other chromatin functions. To test this idea, we used mass spectroscopy to identify H2AX residues additional to serine 139 that are post-translationally modified following exposure of cells to ionizing radiation (IR) and identified several new IR-responsive residues of H2AX. We determined the impact of IR-responsive H2AX residues on cellular resistance to IR and on H2AX-dependent homologous recombination, and also analyzed the contribution to HR of other known or potential post-translationally modified residues of H2AX. The results suggest that the HR and IR-resistance functions of H2AX are controlled in large part by specific MDC1-interacting residues of H2AX, but that additional H2AX residues modulate these core functions of H2AX.
Introduction
Chromosomal DNA double strand breaks (DSBs) provoke an extensive reaction in neighboring chromatin, characterized by phosphorylation of histone H2AX on serine 139, forming “γ-H2AX”.Citation1,Citation2 The major kinase responsible for H2AX phoshorylation is ATM, but other DNA damage response kinases can substitute for ATM.Citation3–Citation6 γ-H2AX, in turn, recruits the chromatin-associated adaptor protein, MDC1,Citation7,Citation8 which interacts with multiple partners, including the Mre11/Rad50/NBS1 (MRN) complex,Citation9 the ATM kinase, and the E3 ubiquitin ligase, RNF8.Citation10–Citation13 RNF8 plays a critical role with RNF168,Citation14,Citation15 in recruiting additional DNA damage response factors to chromatin marked by γ-H2AX. RNF8/RNF168-dependent elements of the γ-H2AX response include 53BP1 and the breast/ovarian cancer predisposition gene products, BRCA1 and BARD1.Citation10–Citation15 Important intermediates in the recruitment of these later factors are the generation of ubiquitinated chromatin species, including ubiquitinated histones H2A and H2AX. Although BRCA1/BARD1 and 53BP1 have known functions in DSB repair—in HR and non-homologous end joining, respectively—each of these exerts its DSB repair functions independently of H2AX.Citation16–Citation19 Indeed, although RNF8 contributes to HR,Citation10,Citation12,Citation13 a major HR function of MDC1 is intact in MDC1 mutants defective for RNF8 binding.Citation18 Further, a recent report indicates that the RNF8/RNF168 pathway contributes to transcriptional silencing at the DSB.Citation20 For these reasons, the biochemical mechanisms of action of γ-H2AX/MDC1 in HR are not yet well understood.
We have taken a genetic approach to the study of γ-H2AX-mediated HR. H2AX null mouse embryonic stem (ES) cells reveal a defect in HR and, specifically, in sister chromatid recombination, that is corrected by either stable or transient expression of wild type (wt) H2AX, but not by expression of H2AX alleles encoding H2AX serine 139 mutants.Citation19 This points to a specific role for γ-H2AX in HR. H2AX tyrosine 142 to alanine (Y142A) mutants, known to be defective for binding to the MDC1 tandem BRCT repeat, were also found to be defective for HR.Citation18 Indeed, the ability of different H2AX Y142 mutants to rescue HR in H2AX null cells correlated with their ability to bind MDC1.Citation18
γ-H2AX is formed in situ in chromatin flanking the chromosomal DSB, and this histone is likely subject to the complex signals that are normally integrated within the chromatin fiber in the form of a “histone code”.Citation21,Citation22 Indeed, ubiquitination of H2A(X) on lysine 118/119, controlled by RNF8 and RNF168, have been implicated in the later stages of the γ-H2AX response, contributing to recruitment of BRCA1/BARD1 (in complex with the Abraxas/Rap80 complex) and 53BP1.Citation10,Citation13,Citation23 Further, the Drosophila melanogaster homolog of H2AX, H2Av, undergoes dTip60-mediated acetylation on lysine 5 as a potential trigger to replace γ-H2Av with unmodified H2Av following DNA repair.Citation24 This points to an intramolecular integration of function within H2Av that controls the dynamics of “γ-H2Av” turnover. Related phenomena have been described in mammalian cells.Citation25 In addition to these DNA damage-specific alterations, H2AX has almost 100% identity with histone H2A throughout the N-terminal tail and core histone fold domains, diverging significantly only at the C-terminal tail. Therefore, it is likely that H2AX is subject to additional non-DNA damage-related post-translational modifications in concert with other H2A species during other chromatin processes.
These observations raised the possibility that mammalian H2AX, like D. melanogaster H2Av, might undergo additional DNA damage-specific post-translational modifications to coordinate its function. Second, it suggested that non-DNA damage related modifications on H2AX might interact functionally with γ-H2AX to modulate DSB repair and HR in particular. To test these ideas, we have used mass spectroscopy to characterize additional IR-induced modifications of H2AX, and have examined the contribution of other post-translationally modified residues of H2AX to H2AX-dependent IR resistance and HR.
Results
IR-induced post-translational modifications of H2AX.
In order to identify additional IR-induced post-translational modifications of histone H2AX, we stably expressed HA-tagged wtH2AX in H2AX−/− mouse embryonic stem (ES) cells, treated these cells with either 0 or 50 Gy of IR, extracted histones with acid, precipitated them with acetone and immunoprecipitated HA-tagged H2AX from the histone extract (). After SDS PAGE (), we used mass spectroscopy to sequence peptides from the anti-HA immunoprecipitates. This revealed several new IR-induced modifications of purified histone H2AX in addition to S139 phosphorylation, as well as certain constitutive post-translational modifications of H2AX. The IR-induced changes included acetylation of K5 and K36 and phoshorylation of T101 (). In addition, T120 and S121 were phosphorylated constitutively (). We did not detect constitutive or IR-induced phosphorylation of Y142, a modification recently identified and associated with apoptosis.Citation26,Citation27 Ubiquitination of K119 of histone H2A and H2AX is well established,Citation23,Citation28 and was again noted ().
Based on previous studies of yeast and mammalian histone H2A, many other residues of histone H2AX are also potential sites for post-translational modifications ( and Suppl. Fig. 1), including S1,Citation29 T123 and T136 phosphorylation,Citation30 K9 acetylation,Citation31,Citation32 and K74, K75 and R76 methylation.Citation33 However, we did not detect these modifications on histone H2AX in this experiment.
IR sensitivity of H2AX mutant cells.
To test whether the above-noted H2AX modifications affect H2AX DSB repair functions, we generated H2AX mutants carrying mutations at relevant residues and expressed each of them in mouse H2AX−/− ES cells. Because H2AX−/− cells show increased IR sensitivity,Citation34,Citation35 we first analyzed whether expression of these H2AX mutants affects IR sensitivity of mouse H2AX−/− ES cells. As expected, complementation with wtH2AX reduced the IR sensitivity of mouse H2AX−/− ES cells (). Like wtH2AX, expression of H2AX mutants K5R, K9R and T136V also reduced IR sensitivity in mouse H2AX−/− ES cells (). This indicates that K5 or K9 acetylation and T136 phosphorylation are dispensable for H2AX-dependent IR resistance, despite the fact that K5 acetylation of H2Av is suggested in the DNA damage response in D melanogaster.Citation24 Recent evidence implicated a role for ubiquitination of H2A and H2AX in the DSB response.Citation23 However, H2AX ubquitination mutant 5KR (Lysine 118, 119, 127, 133 and 134 to arginine), which cannot be ubiquitinated (), complemented the IR sensitivity of mouse H2AX−/− ES cells as effectively as wtH2AX (). This argues against a unique role for H2AX ubiquitination in IR resistance. In contrast, as expected, mouse H2AX−/− ES cells expressing the S139A and Y142A were as sensitive to IR as those expressing the empty vector control. This is consistent with the known roles of S139 phosphorylation and Y142 in recruiting MDC1 during the DSB response.Citation7,Citation8,Citation18 Expression of K36R or T101A did not reverse the IR sensitivity of mouse H2AX−/− ES cells, suggesting that K36 and T101 and, by extension, acetylation of K36 and phosphorylation of T101, participate in H2AX-mediated IR resistance. The steady state abundance of each of these H2AX mutant gene products was comparable with that of wtH2AX ().
S139 phosphorylation of H2AX mutants.
To determine whether the failure of H2AX mutants of K36, T101 and Y142 to reverse IR sensitivity is due to a failure to support S139 phosphorylation of histone H2AX, we transiently transfected mouse H2AX−/− ES cells with H2AX mutants encoding K36R, T101A, Y142A, Y142W or Y142F, in parallel with wtH2AX and empty vector control and analyzed IR-induced S139 phosphorylation for each histone H2AX mutant protein (). With the exception of S139 mutation itself, all of these H2AX mutants displayed appropriate S139 phosphorylation following IR treatment (). S139 phosphorylation of Y142 mutants appeared to be diminished ( and C). However, this likely reflects diminished binding of anti-γ-H2AX antibody to Y142 mutants, as this antibody was developed to target the phosphorylated SQEY peptide and cannot fully recognize the SQE-tail in which Y142 is mutated (unpublished observations). Of note, the ubiquitination levels of these H2AX mutants were not affected ( and C; data not shown), suggesting that these residues do not regulate H2AX ubiquitination.
To determine whether ubiquitination of H2AX regulates S139 phosphorylation or vice versa, we transiently expressed H2AX ubiquitination mutants KK118RR (in which the major ubiquitination target sites, K118 and K119 were each mutated to arginine) and 5KR (in which the five above-noted lysine residues were mutated to arginine) as well as S139A mutant, in parallel, in mouse H2AX−/− ES cells. We found that KK118RR and 5KR were phosphorylated normally on S139 in response to IR (). Further, the S139A mutant showed normal levels of H2AX ubiquitination (). This data suggests that H2AX monoubiquitination and S139 phosphorylation are regulated independently.
We also analyzed monoubiquitination and IR-induced S139 phosphorylation of the following additional H2AX mutants, reflecting sites of known or potential post-translational modification: S1A, K5R, K9R, K74Q, KR75RQ (combined mutations of K75R and R76Q), TS120AV (combined mutations of T120A and S121V) and T136V. These mutants underwent IR-induced S139 phosphorylation and K119 ubiquitination in a manner indistinguishable from wtH2AX ( and B and data not shown), arguing against a role of these residues in regulating these two modifications.
IR-induced focus formation by downstream factors in H2AX mutant cells.
H2AX is required for efficient IR-induced focus formation by MDC1 and by other factors such as 53BP1 and BRCA1/BARD1.Citation36 To determine whether K36, T101, Y142 and ubquitination of H2AX are required for this H2AX function, we stably expressed K36R, T101A, Y142A, Y142W, Y142F and 5KR, in parallel, in mouse H2AX−/− ES cells and analyzed IR-induced focus formation of MDC1 and 53BP1. As expected, in cells containing S139A and Y142A or empty vector control, no IR-induced MDC1 and 53BP1 nuclear foci were observed (). This is consistent with the known inability of H2AX S139A or Y142A mutants to interact with MDC1.Citation7 Unlike Y142A, Y142W and Y142F were able to restore IR-induced MDC1 and 53BP1 nuclear focus formation to mouse H2AX−/− ES cells (). Because neither trypophan nor phenylalanine residues can be phosphorylated, this indicates that Y142 phosphorylation, which has been observed by several groups,Citation26,Citation27 is not required for the recruitment of MDC1 to damaged chromatin. Other H2AX mutants K36R, T101A and 5KR effectively rescued IR-induced focus formation by MDC1 and 53BP1 in mouse H2AX−/− ES cells (; data not shown), suggesting that H2AX K36 acetylation, T101 phosphorylation and ubiquitination of H2AX are not required to set up focus formation of downstream factors in response to IR.
DSB repair by homologous recombination in H2AX mutant cells.
We reported previously that H2AX Y142 as well as S139 are important for its HR function, since H2AX Y142A and S139A are each unable to restore HR to H2AX−/− ES cells.Citation18,Citation19 In contrast, H2AX Y142F and Y142W were shown previously to rescue HR as efficiently as wtH2AX.Citation18 We extended this study to determine whether H2AX K36 acetylation, T101 phosphorylation or ubiquitination of H2AX are important for H2AX-mediated HR. To evaluate the role of K36, T101 and ubiquitination of H2AX in HR, we transiently expressed, in parallel, K36R, T101A, KK118RR and 5KR in mouse H2AX−/− ES cells containing a single copy of an HR/SCR reporter targeted to the ROSA26 locus, with parallel expression of wtH2AX (positive control) and S139A (negative control) and analyzed I-SceI induced HR as described in Materials and Methods. As shown previously,Citation19 HR is less efficient in mouse H2AX−/− ES cells () and wtH2AX, but not H2AX S139A, rescued HR in these cells (). Notably, K36R, T101A and ubiquitination mutants KK118RR and 5KR rescued HR as efficiently as wtH2AX ( and D), suggesting that post-translational modifications of these residues are not required for H2AX-mediated HR. Stable expression of K36R and T101A also rescued HR as effectively as wtH2AX in mouse H2AX−/− HR/SCR reporter ES cells (Suppl. Fig. 2), further confirming that modifications of these two residues are not required for H2AX-dependent HR. Interestingly, stable expression of the H2AX ubiquitination-defective mutant 5KR slightly stimulated HR when compared to wtH2AX, for reasons that are not yet clear (Suppl. Fig. 2).
We further analyzed the ability of H2AX mutants S1A, K5R, K9R, K74R, KR75RQ, T120A, TS120AV and T136V to rescue HR in mouse H2AX−/− HR/SCR reporter ES cells. All of these mutants rescued HR as efficiently as wtH2AX (, D and E), indicating that these residues or modifications occurring at these sites, are not required for H2AX-mediated HR. The effect of Y142 mutation was as noted previously ( and E).
DSB repair by NHEJ in H2AX mutant cells.
We previously developed a rapid reporter for measuring I-SceI-induced NHEJ in mammalian cells.Citation37 This reporter scores positive when a DNA break is induced at two closely positioned, tandem I-SceI sites and repaired by NHEJ. We found that H2AX is dispensable for NHEJ of two closely apposed DNA ends.Citation37 However, modifications on different residues of H2AX might, in theory, offset one another in regulating the action of H2AX in NHEJ, thus possibly masking a role for H2AX in NHEJ. Such interactions could be revealed by specific loss of one NHEJ-regulatory modification, while leaving other modifications intact. To test this possibility, we transiently expressed a panel of H2AX mutants, in parallel, in mouse H2AX−/− ES cells containing a single copy of the NHEJ reporter and analyzed I-SceI-induced NHEJ as described in Materials and Methods. As before, wtH2AX and H2AX S139A served as controls. Consistent with our previous findings, deletion of H2AX had no effect on I-SceI-induced NHEJ () and this was not altered by expression of wtH2AX. Similarly, expression of almost all H2AX mutants did not change NHEJ quantitatively in mouse H2AX−/− ES NHEJ reporter cells (; data not shown). An exception was H2AX Y142A, which appeared to modestly stimulate NHEJ in H2AX−/− ES cells, although the biological significance of this stimulation is not yet clear.
Discussion
The work described here identifies new IR-induced post-translational modifications of H2AX. In addition to serine 139 phosphorylation of H2AX, we identified specific IR-induced acetylation of K5 and K36 and phosphorylation of T101. Interestingly, the K36 mutation, rendering the site unavailable for acetylation, abrogated the ability to reverse IR sensitivity in H2AX−/− cells. Similarly, H2AX T101A mutation, resulting in a site that cannot be phosphorylated on this residue, abolished IR resistance. In contrast, the acetylation defective mutant, K5R, performed equivalently to wtH2AX in reversing IR sensitivity. These results suggest that two of the new modifications identified—K36 acetylation and T101 phosphorylation—contribute to H2AX-mediated IR resistance. Each of these H2AX mutant proteins was incorporated into chromatin and was competent for S139 phosphorylation and for monoubiquitination, performing indistinguishably to wtH2AX in these two regards. Importantly, the ability of each mutant to restore HR to H2AX−/− ES HR reporter cells was also equivalent to that of wtH2AX. This suggests that H2AX residues K36 and T101 contribute to IR resistance by a new, as yet undefined mechanism, which is distinct from the established function of H2AX in HR.Citation18,Citation19 We considered the possibility that the K36R or T101A mutations might have inactivated a previously uncharacterized NHEJ function of H2AX. However, these mutants did not appear to perturb NHEJ of two closely positioned, easily rejoined DSBs. Conceivably, the defect in these mutants has an impact on an IR-resistance function of H2AX that is unrelated to DSB repair or is related to another DSB repair pathway that we have not yet assayed.
The role of H2AX ubiquitination has received significant attention, in view of the coordination of the γ-H2AX/MDC1 response with the recruitment and activation of the E3 ubiquitin ligases, RNF8 and RNF168.Citation10,Citation12–Citation15,Citation20,Citation23,Citation38 Indeed, H2AX ubiquitination might serve as part of the substrate on chromatin that is extensively ubiquitinated during the DNA damage response. We found that mutation of the critical H2AX ubiquitin target lysines, K118 and K119, although it effectively abolished ubiquitination of H2AX, failed to impair the IR resistance function or HR function of H2AX. These results suggest that H2AX ubiquitination is not required for either H2AX-mediated IR resistance or H2AX-mediated HR. Notably, chromatin-associated histone H2A species other than H2AX will retain the capacity to undergo ubiquitination on K118/119. Therefore, our data does not address the recently described role for general H2A ubiquitination in the DNA damage response.Citation23
We have previously analyzed the role of H2AX Y142 in HR and observed a correlation between the ability of specific H2AX Y142 mutants to support HR and their ability to interact with the MDC1 tandem BRCT repeat.Citation18 Consistent with this, H2AX Y142F and Y142W mutants were fully functional for IR resistance and HR in the experiments described here. We were unable to efficiently detect γ-H2AX products of these mutants, likely reflecting loss of affinity of the anti-γ-H2AX antibody for the H2AX C-terminus when it lacks Y142. However, the functionality of the Y142F and Y142W mutants is underscored by their ability to recruit MDC1, 53BP1 and BRCA1 to chromatin in the IR response. The solved crystal structure of the γ-H2AX peptide interacting with the MDC1 tandem BRCT repeat reveals Y142 interacting with a hydrophobic pocket on MDC1.Citation7,Citation8 Presumably, the replacement of Y142 with either F or W residues results in a peptide in which the replacement residues (F or W), which are similarly bulky and hydrophobic, interact with the same hydrophobic pocket. Several recent papers report that H2AX can be phosphorylated on Y142.Citation26,Citation27 We found that the H2AX Y142F mutant, which cannot be phosphorylated, is able to restore both IR resistance and HR as efficiently as wtH2AX, suggesting that Y142 phosphorylation is not required for these DNA damage response functions of H2AX. Indeed, we failed to detect H2AX peptides containing phosphorylated Y142 in the experiments described here. Conceivably, only a small fraction of H2AX is normally phosphorylated on Y142 at a given instant in cycling cells and is dephosphorylated in response to IR. Alternatively, other technical factors might have made the Y142 phosphorylated peptide difficult to detect by mass spectroscopy.
A striking, robust and apparently evolutionarily conserved function of H2AX is its contribution to HR between sister chromatids.Citation18,Citation19,Citation39–Citation41 In vertebrates, this function is mediated by interaction of γ-H2AX with MDC1; however, we noted previously that H2AX/MDC1-mediated HR and BRCA1-mediated HR are genetically separable functions.Citation18,Citation19 Further, MDC1 mutants lacking the ability to recruit RNF8, BRCA1, the MRN complex or 53BP1 to chromatin are nonetheless able to restore HR to MDC1−/− cells. Therefore, the mechanisms by which MDC1 mediates HR remain to be determined. The genetic analysis of H2AX described here underscores the significance of H2AX-mediated HR for the cellular resistance to IR. However, additional DSB repair functions of H2AX are implied by its contribution to class switch recombination (CSR) at IgH loci, a function that is completed by NHEJ.Citation35,Citation42 This suggests that H2AX can mediate both HR and NHEJ, under the appropriate circumstances. However, our attempts to elicit evidence of an NHEJ function of H2AX have thus far yielded no direct evidence in support of this idea.Citation37 In this regard, it may be that H2AX is not required for the rapid religation step of NHEJ—a function that might dominate in the rejoining of two adjacent, easily repaired tandem I-SceI-mediated chromosomal DSBs. In contrast, CSR lesions, which are initiated by interaction of the cytosine deaminase enzyme, AID, with target sites within the IgH locus, may be more complex than those generated by I-SceI.Citation43,Citation44 A further constraint on repair of CSR-associated DSBs is imposed by the extensive (∼100 kb) distance between the DSB ends. Possibly, as the complexity of or distance between the two ends of the break increases, H2AX and its associated proteins might assume a greater functional significance in NHEJ. Indeed, perhaps H2AX mediates a synapsis function that supports HR/SCR under some circumstances and NHEJ under others.
Materials and Methods
Plasmids and antibodies.
The hygromycin resistant expression vectors for HA-tagged human H2AX, S139A and Y142 mutants were described previously.Citation18,Citation19 The other H2AX mutants were generated from the HA-wtH2AX expression plasmid using the QuickChange II site-directed mutagenesis kit (Stratagene). Commercial rabbit polyclonal antibodies used in this study were anti-γ-H2AX (Cat# DR1017) from AbCam, anti-HA-tag (Cat# sc-805) from Santa Cruz Biotechnology, anti-histone H4 (Cat# 06-598) from Upstate and anti-53BP1 (Cat# NB 100-304) from Novus Biologicals. Mouse anti-mouse MDC1 antibody was a gift from Junjie Chen.Citation5
Cell lines, cell culture and transfection.
Isogenic H2AX+/+ and H2AX−/− HR and NHEJ reporter ES clones were generated as described previously.Citation18,Citation19 Transfection of mouse ES cells was performed using Lipofectamine 2000 (Invitrogen) as described previously.Citation19 Mouse H2AX−/− ES cells stably expressing H2AX were generated as described previously.Citation19
Western blotting and immunofluorescence staining.
Histones were acid-extracted and analyzed by western blotting as described previously.Citation19 For immunofluorescence staining, mouse H2AX−/− ES cells stably expressing H2AX and its variants were grown on glass coverslips overnight, irradiated, recovered for 30 minutes, fixed in ice-cold methanol or methanol/acetone, stained with antibodies and imaged by using a Zeiss microscope.
Immunoprecipitation.
Histones were acid-extracted from ∼10Citation8 irradiated mouse H2AX−/− ES cells stably expressing HA-tagged wtH2AX as described previously,Citation19 precipitated in acetone at −20°C overnight, air-dried, dissolved in Lysis Buffer (50 mM Tris-Cl [pH 8.0], 150 mM NaCl, 0.1% NP40, 5 mM NaF, 5 mM Na-Orthovanadate, 5 mM Na-Butyrate, 1 mM PMSF and 1.5 ug/mL protease inhibitor cocktail tablets [Roche]), immunoprecipitated using anti-HA Affinity Matrix (Cat# 1815016, Roche) and resolved by SDS-PAGE electrophoresis. After Coomassie Blue staining, protein bands corresponding to HA-tagged wtH2AX protein were cut from the SDS-PAGE gel and prepared and sent for sequencing by mass spectrometry (Taplin Mass Spectrometry Facility, Harvard Medical School).
I-SceI-induced repair assays.
Analysis of I-SceI-induced GFP+ frequencies in HR or NHEJ reporter mouse ES cells was performed as described previously.Citation18,Citation19,Citation37,Citation45 In co-transfection, each expression plasmid and the I-SceI plasmid were used in equal amounts. Transfection efficiency was measured by parallel transfection of wtGFP expression vector, at an amount one tenth that of the I-SceI expression vector. Statistical analysis was performed with Student's two-tailed paired t-test.
Abbreviations
| 53BP1 | = | p53-binding protein 1 |
| ATM | = | ataxia and telangiectasia mutated |
| BARD1 | = | BRCA1-associated RING domain protein 1 |
| BRCA1 | = | breast cancer predisposition gene 1 |
| BRCT | = | BRCA1 C-terminal domain |
| CSR | = | class switch recombination |
| DSB | = | double strand break |
| ES cells | = | embryonic stem cells |
| HA | = | influenza hemagglutinin peptide |
| HR | = | homologous recombination |
| IP | = | immunoprecipitate |
| IR | = | ionizing radiation |
| MDC1 | = | mediator of DNA damage checkpoint 1 |
| MRN | = | Mre11/Rad50/NBS1 complex |
| MS | = | mass spectroscopy |
| NHEJ | = | non-homologous end joining |
| RNF8 | = | RING finger protein 8 |
| RNF168 | = | RING finger protein 168 |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| Ub | = | ubiquitin |
| wt | = | wild type |
Figures and Tables
Figure 1 Strategy for identification of IR-induced post-translational modifications of H2AX. (A) Outline of protocol for identifying IR-induced post-translational modifications of H2AX. IP: immunoprecipitation; HA: influenza hemagglutinin peptide; MS: mass spectroscopy. (B) Coomassie blue staining of immunoprecipitates after SDS-PAGE analysis. Several histone species are shown. FT: flow-through. uH2A: monoubiquitinated histone H2A. “*” indicates the HA-H2AX bands, which were cut from the gel and purified for analysis by MS.
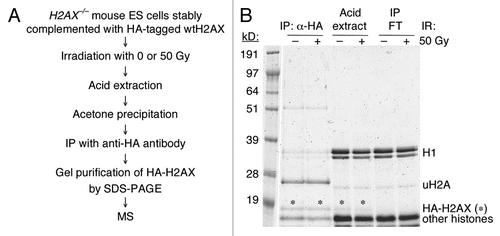
Figure 2 IR-sensitivity of H2AX−/− ES cells expressing H2AX mutants. (A) Survival rate of H2AX−/− ES cells expressing individual H2AX mutants after exposure to IR. Each survival rate, normalized to transfection efficiency and plating efficiency, represents mean of triplicate samples. Error bars indicate standard error of the mean (s.e.m.). (B) Steady-state level of HA-tagged H2AX mutants expressed in H2AX−/− ES cells. Treatment with 10 Gy of IR is indicated. γ-H2AX, HA-H2AX and HA-uH2AX as shown are detected by anti-γ-H2AX and anti-HA antibodies respectively.
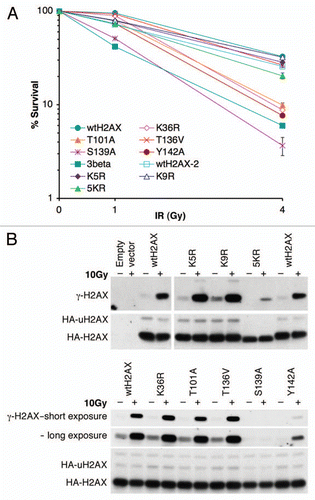
Figure 3 S139 phosphorylation and K119 monoubiquitination of H2AX mutants. H2AX−/− ES cells were transiently transfected with mammalian expression vectors encoding H2AX mutants shown and treated with IR as indicated 3 days after transfection. 30 minutes post-IR, histones were extracted for analysis by western blotting. γ-H2AX, HA-H2AX and HA-uH2AX are shown. Wild type H2AX (wtH2AX), S139A and empty vector were used as controls in each experiment. H2AX mutants DeIN (deletion of N-terminal 15 residues of H2AX), K5R, K9R and K36R are grouped in (A). K74Q, K75Q, KR75RQ (K75 to R and R76 to Q), T101A, 5KR (K118, 119, 127, 133 and 134 to R) and T136V as well as Y142A are grouped in (B). KK118RR (K118 and 119 to R), Y142F and Y142W as well as 5KR and Y142A are grouped in (C).
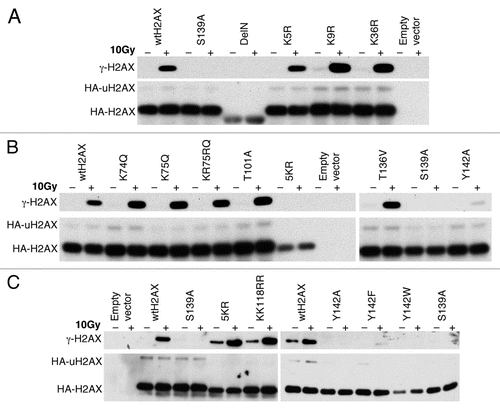
Figure 4 Effect of H2AX mutations on IR-induced focus formation of MDC1 and 53BP1. H2AX−/− ES cells were transiently transfected, in parallel, with mammalian expression vectors encoding H2AX Y142 mutants or ubiquitination mutant 5KR and then treated with 10 Gy of IR. Cells were immunostained for HA-H2AX species, MDC1 and 53BP1. DAPI staining is also shown.

Figure 5 Effect of H2AX mutations on H2AX-dependent HR. Percentage of I-SceI-induced GFP+ cells from H2AX+/+ and H2AX−/− HR reporter mouse ES cells (A) or H2AX−/− HR reporter mouse ES cells transiently transfected with mammalian expression vectors encoding H2AX mutants as indicated in (B–E). See Materials and Methods for details. Bars represent mean of triplicates. Error bars indicate s.e.m.
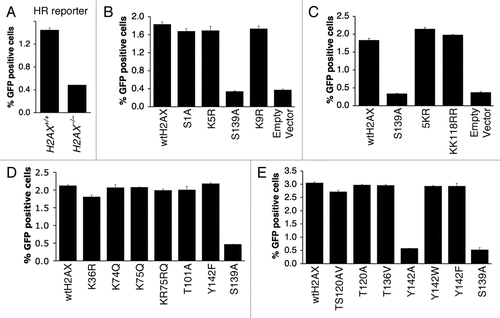
Figure 6 Effect of H2AX mutations on I-SceI-induced NHEJ. Percentage of I-SceI-induced GFP+ cells from H2AX+/+ and H2AX−/− NHEJ reporter mouse ES cells (A) or H2AX−/− HR reporter mouse ES cells transiently transfected with mammalian expression vectors encoding H2AX mutants as indicated in (B). Bars represent mean of triplicates. Error bars indicate s.e.m.
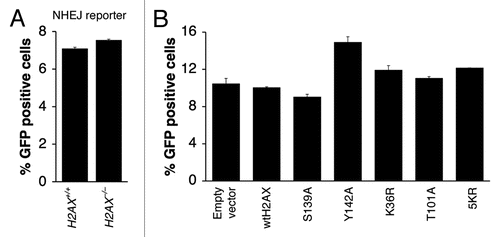
Table 1 Identification of post-translational modifications of HA-H2AX by MS
Additional material
Download Zip (1.1 MB)Acknowledgements
We thank Drs. Frederick Alt, Craig Bassing and Stephen Jackson, as well as members of the Scully lab, for insightful comments and helpful discussions. Dr. Junjie Chen kindly provided antibodies as noted in the text. We thank Dr. Steven Gygi and members of the Taplin Mass Spectrometry Facility, Harvard Medical School, for analysis of H2AX peptides. This work was supported by NIH grants GM073894, CA095175 and a Leukemia and Lymphoma Society Scholar Award (to R.S.).
References
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905 - 916
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858 - 5868
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462 - 42467
- Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 2001; 276:47759 - 47762
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 2006; 21:187 - 200
- Savic V, Yin B, Maas NL, Bredemeyer AL, Carpenter AC, Helmink BA, et al. Formation of dynamic gammaH2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol Cell 2009; 34:298 - 310
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213 - 1226
- Lee MS, Edwards RA, Thede GL, Glover JN. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gammaH2AX histone tail. J Biol Chem 2005; 280:32053 - 32056
- Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep 2008; 9:795 - 801
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901 - 914
- Kim H, Chen J. New players in the BRCA1-mediated DNA damage responsive pathway. Mol Cells 2008; 25:457 - 461
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007; 318:1637 - 1640
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887 - 900
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435 - 446
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009; 136:420 - 434
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell 1999; 4:511 - 518
- Scully R, Livingston DM. In search of the tumoursuppressor functions of BRCA1 and BRCA2. Nature 2000; 408:429 - 432
- Xie A, Hartlerode A, Stucki M, Odate S, Puget N, Kwok A, et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol Cell 2007; 28:1045 - 1057
- Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, et al. Control of sister chromatid recombination by histone H2AX. Mol Cell 2004; 16:1017 - 1025
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010; 141:970 - 981
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41 - 45
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074 - 1080
- Panier S, Durocher D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair 2009; 8:436 - 443
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR 3rd, et al. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 2004; 306:2084 - 2087
- Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol 2007; 27:7028 - 7040
- Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, et al. WSTF regulates the H2AX DNA damage response via a novel tyrosine kinase activity. Nature 2009; 457:57 - 62
- Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009; 458:591 - 596
- Bohm L, Crane-Robinson C, Sautiere P. Proteolytic digestion studies of chromatin core-histone structure. Identification of a limit peptide of histone H2A. Eur J Biochem 1980; 106:525 - 530
- Barber CM, Turner FB, Wang Y, Hagstrom K, Taverna SD, Mollah S, et al. The enhancement of histone H4 and H2A serine 1 phosphorylation during mitosis and S-phase is evolutionarily conserved. Chromosoma 2004; 112:360 - 371
- Moore JD, Yazgan O, Ataian Y, Krebs JE. Diverse roles for histone H2A modifications in DNA damage response pathways in yeast. Genetics 2007; 176:15 - 25
- Goll MG, Bestor TH. Histone modification and replacement in chromatin activation. Genes Dev 2002; 16:1739 - 1742
- Turner BM. Cellular memory and the histone code. Cell 2002; 111:285 - 291
- Zhang L, Eugeni EE, Parthun MR, Freitas MA. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma 2003; 112:77 - 86
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of his-tone H2AX. Proc Natl Acad Sci USA 2002; 99:8173 - 8178
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, et al. Genomic instability in mice lacking histone H2AX. Science 2002; 296:922 - 927
- Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 2006; 5:534 - 543
- Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol 2009; 16:814 - 818
- Bekker-Jensen S, Rendtlew Danielsen J, Fugger K, Gromova I, Nerstedt A, Lukas C, et al. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat Cell Biol 2010; 12:80 - 86
- Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, et al. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol 2004; 14:1703 - 1711
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, et al. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell 2004; 16:991 - 1002
- Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell 2004; 16:1003 - 1015
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell 2006; 21:201 - 214
- Dudley DD, Chaudhuri J, Bassing CH, Alt FW. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol 2005; 86:43 - 112
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Ann Rev Biochem 2010; 79:181 - 211
- Nagaraju G, Odate S, Xie A, Scully R. Differential regulation of short- and long-tract gene conversion between sister chromatids by Rad51C. Mol Cell Biol 2006; 26:8075 - 8086