
Abstract
Our recent studies have mechanistically implicated a loss of stromal Cav-1 expression and HIF1-alpha-activation in driving the cancer-associated fibroblast phenotype, through the paracrine production of nutrients via autophagy and aerobic glycolysis. However, it remains unknown if HIF1a-activation is sufficient to confer the cancer-associated fibroblast phenotype. To test this hypothesis directly, we stably-expressed activated HIF1a in fibroblasts and then examined their ability to promote tumor growth using a xenograft model employing human breast cancer cells (MDA-MB-231). Fibroblasts harboring activated HIF1a showed a dramatic reduction in Cav-1 levels and a shift towards aerobic glycolysis, as evidenced by a loss of mitochondrial activity, and an increase in lactate production. Activated HIF1a also induced BNIP3 and BNIP3L expression, markers for the autophagic destruction of mitochondria. Most importantly, fibroblasts expressing activated HIF1a increased tumor mass by ~2-fold and tumor volume by ~3-fold, without a significant increase in tumor angiogenesis. In this context, HIF1a also induced an increase in the lymph node metastasis of cancer cells. Similar results were obtained by driving NFκB activation in fibroblasts, another inducer of autophagy. Thus, activated HIF1a is sufficient to functionally confer the cancer-associated fibroblast phenotype. It is also known that HIF1a expression is required for the induction of autophagy in cancer cells. As such, we next directly expressed activated HIF1a in MDA-MB-231 cells and assessed its effect on tumor growth via xenograft analysis. Surprisingly, activated HIF1a in cancer cells dramatically suppressed tumor growth, resulting in a 2-fold reduction in tumor mass and a 3-fold reduction in tumor volume. We conclude that HIF1a activation in different cell types can either promote or repress tumorigenesis. Based on these studies, we suggest that autophagy in cancer-associated fibroblasts promotes tumor growth via the paracrine production of recycled nutrients, which can directly "feed" cancer cells. Conversely, autophagy in cancer cells represses tumor growth via their "self-digestion." Thus, we should consider that the activities of various known oncogenes and tumor-suppressors may be compartment and cell-type specific, and are not necessarily an intrinsic property of the molecule itself. As such, other "classic" oncogenes and tumor suppressors will have to be re-evaluated to determine their compartment specific effects on tumor growth and metastasis. Lastly, our results provide direct experimental support for the recently proposed "Autophagic Tumor Stroma Model of Cancer."
Acknowledgements
M.P.L. and his laboratory were supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660) and the Susan G. Komen Breast Cancer Foundation. F.S. was supported by grants from the Breast Cancer Alliance (BCA) and a Research Scholar Grant from the American Cancer Society (ACS). R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
We also thank Drs. William C. Hahn (Harvard/Dana Farber) and William G. Kaelin (Harvard/Dana Farber) for generously providing the necessary plasmid vectors encoding IKBKE, HIF1α (wild-type) and HIF1α (mutationally-activated).
Figures and Tables
Figure 1 Expression of activated HIF1a or IKBKE in fibroblasts downregulates Cav-1 protein expression. (A) Generating stable fibroblast cell lines expressing HIF1a. hTERT-BJ1 fibroblasts were transduced with retroviral vectors harboring HIF1a (wt, wild-type; or mut, mutationally activated) or the pBABE vector alone as a negative control. After selection for puromycin resistance, the “pool” was subjected to immuno-blot analysis with a battery of antibody probes. HIF1a expression was confirmed using antibodies directed against HIF1a, as well as the HA-epitope tag. Cav-1 expression was monitored with antibodies directed against total Cav-1 and tyrosine-phosphorylated Cav-1 (pY14). Note that activated HIF1a downregulates total Cav-1 levels, but increases the relative levels of phospho-Cav-1. In contrast, wild-type HIF1a only increases the levels of phospho-Cav-1. Immunoblotting with beta-actin is shown as a control for equal loading. (B) Generating stable fibroblast cell lines expressing IKBKE. hTERT-BJ1 fibroblasts were transduced with IKBKE, alone or in combination with activated HIF1a. See the Materials and Methods section for more details on how the double-transfectant was generated. IKBKE expression was confirmed using antibodies directed against NFκB (p65), as well as the FLAG-epitope tag. Cav-1 expression was monitored with antibodies directed against total Cav-1. Note that IKBKE downregulates total Cav-1 levels, but that co-expression of IKBKE and activated HIF1a reverts this phenotype. Immunoblotting with beta-actin is shown as a control for equal loading.
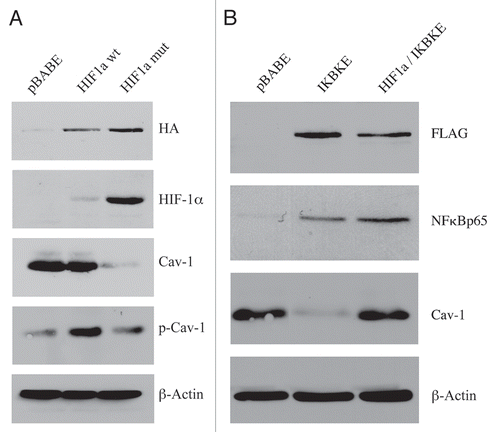
Figure 2 Chloroquine rescues the expression of phospho-Cav-1 (pY14) in activated HIF1a and IKBKE transfected fibroblasts. hTERT-BJ1 fibroblasts recombinantly expressing activated HIF1a or IKBKE were incubated with chloroquine for 24 or 48 hours and then subjected to immunoblot analysis with antibodies directed against phospho-Cav-1 (pY14). Note that treatment with chloroquine rescues the expression of phospho-Cav-1 (pY14) in both fibroblast cell lines. Immunoblotting with beta-actin is shown as a control for equal loading.
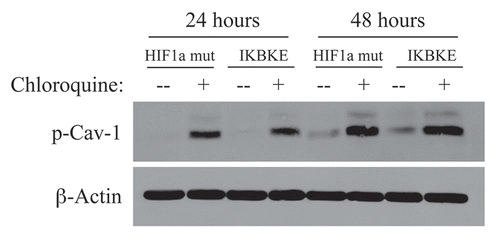
Figure 3 Reductions in mitochondrial respiratory chain complexes in activated HIF1a and IKBKE transfected fibroblasts. To assess the status of the mitochondrial respiratory chain, fibroblast lysates were prepared and subjected to immunoblot analysis with a battery of antibodies directed against mitochondrial complex components (I–V). Note that expression of activated HIF1a reduces components of mitochondrial complex I, II, III and IV, while expression of IKBKE reduces components of complex I and IV. However, co-expression of activated HIF1a and IKBKE reverts this phenotype. Immunoblotting with beta-actin is shown as a control for equal loading.

Figure 4 BNIP3L protein expression is strongly induced in HIF1a and IKBKE transfected fibroblasts. BNIP3L is a marker of mitochondrial autophagy, i.e., mitophagy, which is transcriptionally-induced by HIF1a activation. Note that expression of HIF1a (wild-type or mutationally-activated) or IKBKE induces BNIP3L expression. However, co-expression of activated HIF1a and IKBKE reverts this phenotype. Immunoblotting with beta-actin is shown as a control for equal loading.
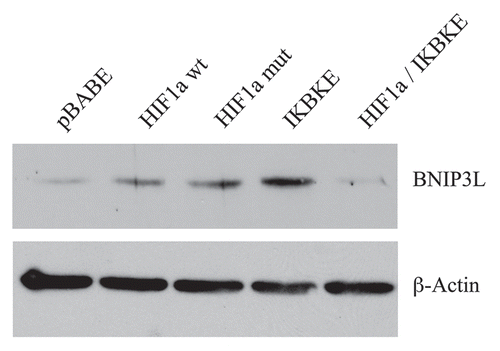
Figure 5 Localization of mitophagy markers (BNIP3 and BNIP3L) in activated HIF1a transfected fibroblasts. BNIP3 and BNIP3L are two distinct genes which are markers of mitophagy, and are thought to be under the transcriptional control of HIF1a activation. Note that both BNIP3 (A) and BNIP3L (B) are upregulated in fibroblasts harboring activated HIF1a, as compared with the vector alone control (pBABE). Both show a morphological pattern that is consistent with mitochondrial and/or autophagosomal localization, as expected.
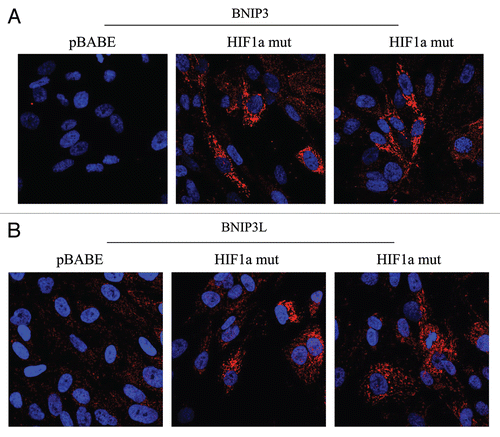
Figure 6 Localization of LC3, an autophagy marker, in activated HIF1a transfected fibroblasts. LC3 is a well-established marker of autophagosomes. Note that LC3 immunostaining yields a characteristic fluorescence pattern that is consistent with the autophagic phenotype, in fibroblasts expressing activated HIF1a.
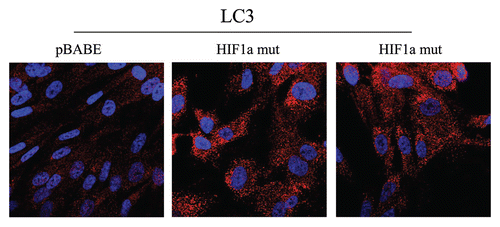
Figure 7 Lactate accumulation in activated HIF1a transfected fibroblasts. hTE RT fibroblasts expressing HIF1a (wild-type or mutationally activated) or IKBKE were grown in tissue culture media. Then, the media was harvested and subjected to a biochemical analysis to determine its lactate content. Note that fibroblasts expressing activated HIF1a secrete the most lactate (p ≤ 0.02), as expected. Similarly, fibroblasts expressing IKBKE also secrete more lactate, although it did not reach statistical significance. A red line indicates baseline levels of lactate relative to HIF1a wild-type.
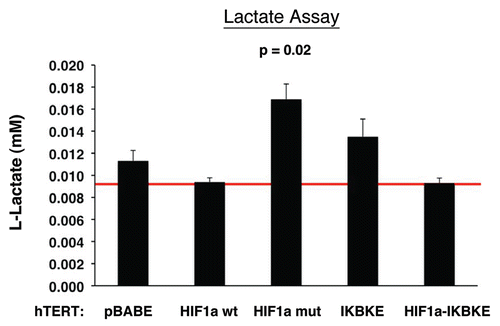
Figure 8 Fibroblasts harboring activated HIF1a promote mitochondrial activity in adjacent cancer cells. MitoTracker is used to visualize functional mitochondria with an active membrane potential. (A) Fibroblasts alone. Note that fibroblasts harboring activated HIF1a show reduced MitoTracker staining, consistent with a decrease in mitochondrial mass and/or membrane potential. (B) Co-culture. Note that fibroblasts harboring activated HIF1a increase the MitoTracker staining profile of adjacent MDA-MB-231 cancer cells, when they are co-cultured. MDA-MB-231 breast cancer cells are marked by GFP expression.
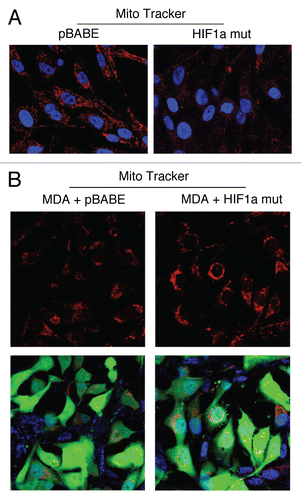
Figure 9 Visualization of peri-nodal metastatic breast cancer cells using GFP. Many of the mice co-injected with fibroblasts (GFP-negative) and cancer cells (GFP-positive) showed lymph node involvement, characterized by enlarged lymph nodes. To validate that this phenomenon was indeed related to lymph node metastasis, tissue sections were cut and used to detect the presence of GFP-positive cancer cells by GFP auto-fluorescence. Note the presence of GFP-positive cancer cells in the peri-nodal fat. Several examples are shown. Nuclei were stained with DAPI (blue color).
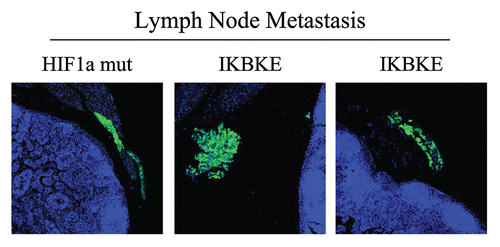
Figure 10 Fibroblasts harboring activated HIF1a increase breast cancer tumor growth, without a significant increase in angiogenesis: 4 weeks post-injection. (A and B) Quantitation of tumor mass and volume. Note that the expression of activated HIF1a in fibroblasts substantially promoted tumor growth, resulting in an ∼2-fold increase in tumor mass and a near 3-fold increase in tumor volume. Similarly, fibroblasts expressing wild-type HIF1a or IKBKE also promoted tumor growth, but activated HIF1a showed the largest tumor promoting activity. Conversely, co-expression of activated HIF1a and IKBKE in fibroblasts abrogated this tumor promoting activity, as expected based on our in vitro phenotypic analysis. p values are as shown. CTR, represents control. As a control, we injected MDA-MB-231 cells alone or in combination with hTERT-BJ1 fibroblasts transfected with the vector alone (pBABE). Since no significant differences between these 2 control groups were observed, they were combined (N = 19 tumors for this control group). For all the other experimental groups, N = 10 tumors per group. (C) Quantitation of tumor angiogenesis. Frozen sections from the tumors were cut and immuno-stained with anti-CD31 antibodies and vessel density was quantitated. Note that no significant increases in vessel density were observed, suggesting that the tumor promoting effects of the transfected fibroblasts we observe are independent of angiogenesis.
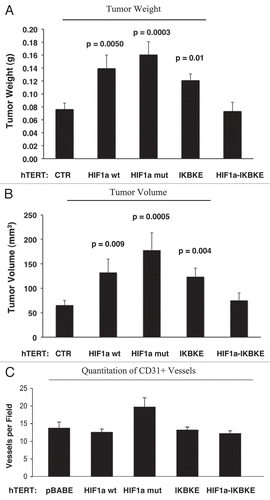
Figure 11 Fibroblasts harboring activated HIF1a increase breast cancer tumor growth: 2 weeks post-injection. The tumor promoting effects of fibroblasts harboring activated HIF1a were also assessed at 2 weeks post tumor cell injection. CTR, represents control. As a control, we injected MDA-MB-231 cells alone or in combination with hTERT-BJ1 fibroblasts transfected with the vector alone (pBABE). Since no significant differences between these 2 control groups were observed, they were combined (N = 20 tumors for this control group). For all the other experimental groups, N = 10 tumors per group. Note that the tumor promoting effects of fibroblasts expressing activated HIF1a are still evident earlier on in tumorigenesis, with significant increases in tumor mass and volume (A), but no significant increases in angiogenesis were observed (B). p values are as shown. n.s., not significant.
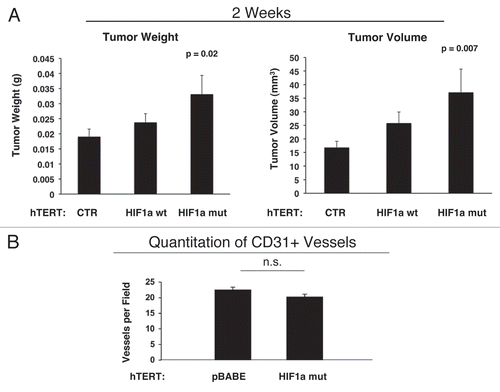
Figure 12 Activated HIF1a downregulates Cav-1 in the tumor stromal compartment: Stromal co-localization with PKM2 and LDHB. (A) Cav-1 immunostaining. To assess whether expression of activated HIF1a in fibroblasts results in mammary tumors with a Cav-1 negative fibroblastic stromal compartment, consecutive frozen sections were cut from mammary tumors that were grown with fibroblasts expressing activated HIF1a. Note that Cav-1 expression is substantially reduced in the stromal compartment of these tumors, but is selectively retained in the micro-vasculature (endothelial cells), exactly as is observed in human breast cancers in vivo. Tumor cells are marked by GFP (green fluorescence). (B) PKM2 and LDHB immunostaining. The presence of these Cav-1 negative fibroblasts was confirmed in consecutive sections (compare with part A, shown at right) by employing antibodies directed against glycolytic enzymes, such as PKM2 and LDHB. Note that PKM2 and LDHB immuno-staining nicely coincides with the Cav-1 negative stromal area in these MDA-MB-231 tumor tissue sections, and does not significantly overlap with the tumor cells which are marked by GFP (green fluorescence).
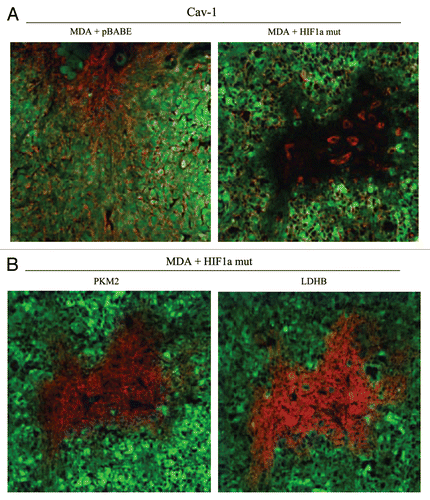
Figure 13 Expression of activated HIF1α in human breast cancer cells suppresses tumor growth and induces autophagy. (A) Quantitation of tumor mass and volume. We hypothesized that HIF1a expression in cancer cells would retard tumor growth, due to the “self-digestion” of the tumor cells. To test this hypothesis, MDA-MB-231 cells were injected into the flanks of nude mice. Two-weeks post-injection, tumors were harvested. Note that the expression of activated HIF1a in MDA-MB-231 tumor cells significantly retards tumor growth, resulting in >2-fold reduction in tumor mass and a >3-fold reduction in tumor volume. p values are as shown. For all the experimental groups, N = 10 tumors per group. (B) Activated HIF1α in human breast cancer cells downregulates Cav-1 protein expression. To study the possible compartment-specific effects of HIF1a activation on breast cancer tumor growth, we also directly expressed HIF1a (wt or mutationally activated) in MDA-MB-231 cells. Cav-1 expression was monitored with antibodies directed against total Cav-1. Note that activated HIF1a downregulates total Cav-1 levels. Immunoblotting with beta-actin is shown as a control for equal loading. (C) Localization of LC3, an autophagy marker, in activated HIF1α transfected breast cancer cells. LC3 is a well-established marker of autophagy. Note that LC3 immuno-staining yields a characteristic fluorescence pattern that is consistent with the autophagic phenotype, in human breast cancer cells expressing activated HIF1a.
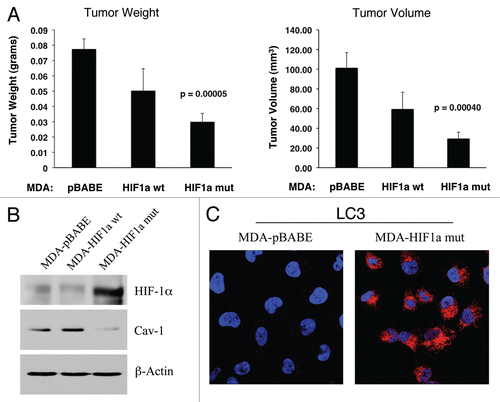
Figure 14 Activated HIF1a in Human Breast Cancer Cells Does Not Appear to Induce Mitophagy. (A) Mitochondrial respiratory complexes. To assess the status of the mitochondrial respiratory chain, breast cancer cell lysates were prepared and subjected to immunoblot analysis with a battery of antibodies directed against mitochondrial complex components (I–V). Note that expression of activated HIF1a does not affect components of mitochondrial complexes I–V. Immunoblotting with beta-actin is shown as a control for equal loading. (B) BNIP3L expression. Note that expression of HIF1a (wild-type or mutationally-activated) does not affect BNIP3L expression. Immunoblotting with beta-actin is shown as a control for equal loading.
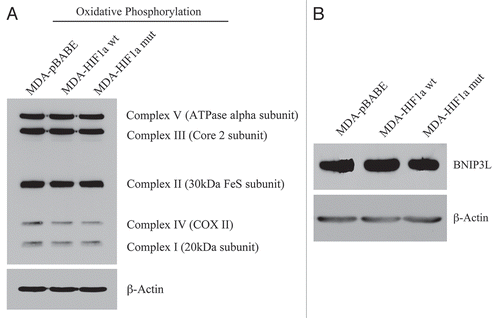
Figure 15 Autophagic cancer associated fibroblasts promote tumor growth, via the paracrine production of recycled nutrients. Previously, we demonstrated that cancer cells induce oxidative stress in adjacent fibroblasts, thereby conferring an autophagic cancer associated fibroblast (CAF) phenotype. This results in the autophagic destruction of of caveolae (Cav-1) and mitochondria, which produces recycled nutrients via aerobic glycolycis (such as lactate) that can then be transferred to cancer cells, to stimulate tumorigenesis, via oxidative phosphorylation (“the reverse Warburg effect”). Similarly, loss of stromal Cav-1 is sufficient to promote more oxidative stress, via increased NO production and the resulting mitochondrial dysfunction.Citation13,Citation15 Here, we show that we can mimic this process simply by overexpressing activated HIF1a (highlighted in blue) in the stromal fibroblast compartment. Similar results were obtained with NFκB-activation, which also is sufficient to drive the induction of autophagy/mitophagy. Thus, we suggest that catabolism in the fibroblasts “fuels” the anabolic growth of adjacent epithelial cancer cells, via “recycled” nutrients. ROS, reactive oxygen species; NO, nitric oxide.
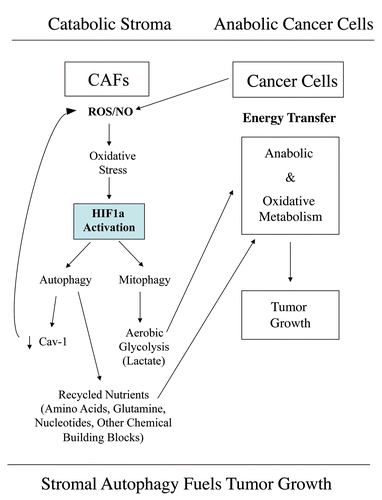
Figure 16 The functional activity of tumor suppressors and oncogenes may be cell-type and compartment specific: HIF1a as a key example. Here, we have shown that activated HIF1a behaves as a tumor promoter in cancer-associated fibroblasts (CAFs) and as a tumor suppressor in epithelial cancer cells. Thus, tumor suppressor or oncogenic activity may not be an intrinsic property of a given molecule per se, but where it is expressed may determine the functional consequences and clinical outcome. As such, the functional activity of “classic” tumor suppressors and oncogenes is compartment and cell-type specific. Horizontal arrows indicate the presumed direction of energy transfer (via Recycled Nutrients) based on autophagy induced by the expression of activated HIF1a. HIF1+, denotes HIF1-alpha activation/stabilization.
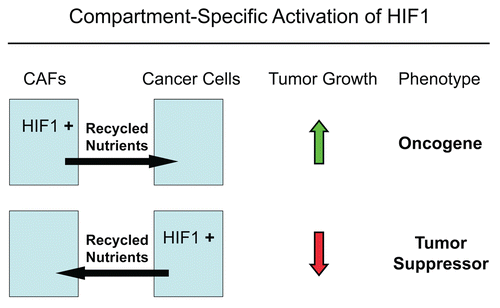
Table 1 Proteomic analysis of fibroblasts harboring activated HIF1α
Table 2 Proteomic analysis of IKBKE-transfected fibroblasts
Table 3 Tumor incidence: hTERT fibroblasts alone do not form tumors