Abstract
Development of the cerebellum, a brain region regulating posture and coordination, occurs post-natally and is marked by rapid proliferation of granule neuron precursors (CGNPs), stimulated by mitogenic Sonic hedgehog (Shh) signaling. β-Arrestin (βArr) proteins play important roles downstream of Smoothened, the Shh signal transducer. However, whether Shh regulates βArrs and what role they play in Shh-driven CGNP proliferation remains to be determined. Here, we report that Shh induces βArr1 accumulation and localization to the nucleus, where it participates in enhancing expression of the cyclin dependent kinase (cdk) inhibitor p27, whose accumulation eventually drives CGNP cell cycle exit. βArr1 knock-down enhances CGNP proliferation and reduces p27 expression. Thus, Shh-mediated βArr1 induction represents a novel negative feedback loop within the Shh mitogenic pathway, such that ongoing Shh signaling, while required for CGNPs to proliferate, also sets up a cell-intrinsic clock programming their ultimate exit from the cell cycle.
Acknowledgements
We thank Betsy Ross and her lab members for helpful discussion in preparation of this manuscript. Marc Caron kindly provided βArr1 plasmids. These studies were funded by NRSA F32 AG030888 from the National Institute on Aging (Susana Parathath) and NIH R01 NS061070 (Anna Marie Kenney).
Figures and Tables
Figure 1 Shh upregulates βArr1 protein, phosphorylation and nuclear localization. (A) CGNPs were treated with or without Shh as indicated for 48 hours, and lysates were blotted for βArr1, PSer412-βArr1, βArr2 and cyclin D2. Tubulin demonstrates equal protein loading. (B) Quantification of (A). Average fold change for each protein was calculated by normalization to Tubulin levels. Each Shh-mediated fold change was further normalized to the corresponding vehicle sample. βArr1, PSer412-βArr1 and cyclin D2 are significantly increased in Shh treated CGNPs. (C) RNA isolated from vehicle or Shh treated CGNPs was analyzed by qRT-PCR to evaluate βArr1 expression. Shh treatment does not induce βArr1 mRNA as indicated by fold change over β-actin control. (D) CGNPs treated with or without Shh for 24 h prior to treatment with 1 µg/mL cyclopamine. Cells were fixed and stained for βArr1 (red) and nuclei were visualized by DAPI (blue). βArr1 is primarily cytoplasmic in vehicle-treated (left part and inset) and Shh + cyclopamine-treated CGNPs (right part and inset) compared to Shh-treated cells (center part and inset). (E) Percentage of cells expressing βArr1 and percentage of those with nuclear βArr1 from staining in (D) (n > 300 cells in 3 or more fields). There is a significant increase in the number of cells with nuclear βArr1 in the Shh-treated group. (F) Sub-cellular fractionation followed by western blot analysis of vehicle, Shh or Shh plus cyclopamine-treated CGNPs to confirm increased nuclear localization of βArr1 with Shh. In the presence of cyclopamine, total and phospho-βArr1 levels in the nucleus decrease. cJun/Ap1 was used as nuclear protein control and GAPDH as cytoplasmic control. See also Supplemental Figure 1 for verification of βArr1 antibody specificity and analysis of Smoothened antagonist on βArr1.
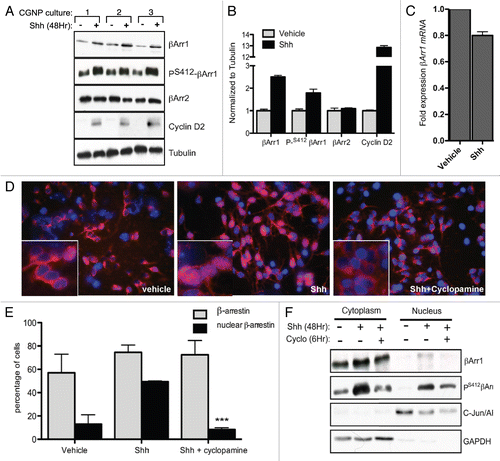
Figure 2 βArr1 knockdown increases Shh-mediated CGNP proliferation while overexpression reduces Ki67 and cyclin D2 levels. (A) CGNPs were infected with a pool of lentiviruses containing sequences targeting βArr1. Cells were fixed, stained for Ki67 and DAPI, and the number of Ki67-positive cells was counted (graph). Lysates were analyzed by western blot to demonstrate βArr1 knockdown and increased cyclin D2 levels. (B) CGNPs were infected with retroviruses containing FLAG-tagged βArr1 and treated with Shh for 48 hours. Cells were fixed, stained for Ki67 and DAPI, and the number of Ki67-positive cells was determined (graph). Lysates were analyzed by western blot to show βArr1 overexpression, as determined by FLAG levels and reduced cyclin D2 levels. Cleaved caspase-3 levels indicate that reduced proliferation is not a result of increased apoptosis. (C) Infection of cerebellar slices with lentiviruses targeting βArr1 causes increased Ki67 staining (red) in the EGL (designated by white line). **/++ or *** indicates statistically significant difference compared to vehicle treated CGNPs (p < 0.005 and p < 0.001 respectively, n = 5). See also Supplemental Figure 2 for analysis of control infections.
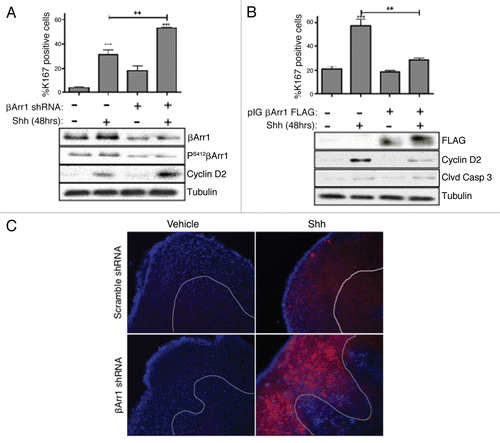
Figure 3 βArr1 phosphorylation at Ser412 is necessary and sufficient to mediate βArr1 accumulation in the nucleus. (A) CGNPs were treated with Shh for 48 h prior to treatment with okadaic acid (OA, 100 nM) for 3 hrs. Western blot analysis shows that OA treatment increases phosphorylation of βArr1 at Ser412, even in the absence of Shh. (B) Treatment of CGNPs with OA for 3 h mediates the translocation of endogenous βArr1 (red) from the cytoplasm to the nucleus in both vehicle and Shh treated cells. Nuclei were visualized with DAPI (blue). (C) CGNPs were infected with βArr1 FLAG retroviruses WT or Ser412 phospho-mutants (S412A or S412D) and were treated with Shh as indicated for 48 h. Cells were then immuno-stained with antibodies against FLAG (red), BrdU (green) and DAPI (blue). Ser-Ala (S412A) mutation prevents βArr1 nuclear localization in Shh treated CGNPs, as determined by FLAG-positive cells (red, middle parts). However, Ser-Asp (S412D) mutants show exclusively nuclear βArr1 in both vehicle and Shh treated CGNPs (arrowheads, right parts). (D) CGNPs infected with wild-type and mutant βArr1 retroviruses were collected for western blot analysis to evaluate FLAG, cyclin D2 and β-tubulin levels. Cyclin D2 is reduced in FLAG βArr1 wild-type- and S412D-infected cells. (E) CGNPs were pulsed with BrdU for 8 h prior to fixation and staining. Cells infected with either βArr1 WT or βArr1 Ser-Asp mutation (βArr1S412D) show reduced proliferation in response to Shh. ** or ***indicates statistically significant difference to vehicle treated CGNPs (p < 0.005 and p < 0.001 respectively, n = 3).
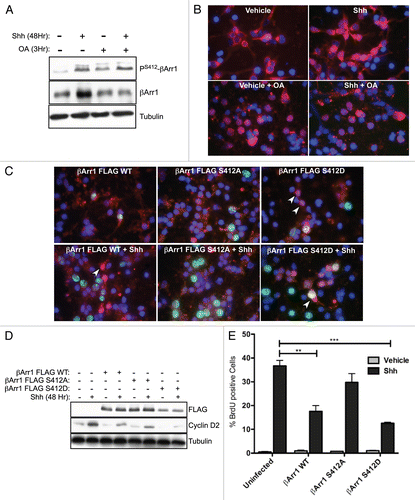
Figure 4 βArr1, p27 and PS412-βArr1 co-localize in vivo and in vitro. (A) Serial sections from PN day 0, 7 and 15 WT mice were stained for either p27, βArr1 or PS412-βArr1, the indicated proteins as described in the methods. Expression of p27 can be seen in the post-mitotic CGNP layer (EGLb) from PN0 and most clearly at PN7. Lower levels of p27 are seen in the proliferative layer of the EGL (EGLa), higher magnification images. βArr1 and PS412-βArr1 are most highly expressed at PN7, with peak nuclear localization occurring in the EGLb (black arrows, higher magnification images). They are also expressed in the remaining EGL of PN15 mice but not in the IGL. (B) CGNPs were cultured in the presence of Shh for 48 h before fixation and stained with βArr1 (red) and p27 (green). Nuclei were visualized by DAPI (blue). Arrowheads indicate cells with nuclear co-localization of βArr1 with p27. See also Supplemental Figure 3 for in vivo analysis of CREB and P-CREB.
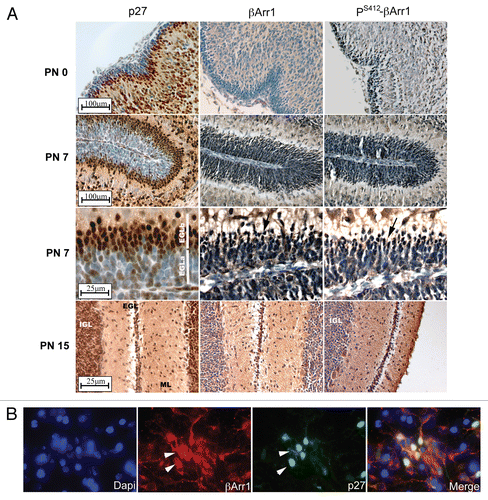
Figure 5 Nuclear βArr1 regulates p27 levels. (A) Time course of CGNPs treated with Shh for increasing periods of time shows increased p27 and βArr1 protein levels by western blotting. PS412-βArr1 levels also increase over time, correlating to the Shh- and time-dependent increase in Gli1 transcription (graph). Tubulin is shown as a loading control. (B) RNA was isolated from CGNPs and treated with Shh for the indicated time points and qPCR performed to evaluate p27 expression. Expression level was normalized to that in vehicle-treated CGNPs. p27 mRNA levels increase over time in the presence of Shh, leveling off after 72 h. (C) CGNPs treated with Shh for 48 h were immunostained for p27, and the ratio of p27-positive cells to DAPI-only (i.e., p27-negative) cells was determined; there is no change in the overall number of cells expressing p27. (D and E) CGNPs were infected with lentiviruses expressing shRNAs targeting βArr1 and the effects on p27 were determined. βArr1 knockdown is associated with reduced p27 protein (western blot, D) and mRNA (graph, E). (F) Western blot analysis of CGNPs transduced with βArr1 FLAG retroviruses demonstrates increased p27 levels. * or ***indicates statistically significant difference compared to vehicle treated CGNPs (p < 0.05 and p < 0.001 respectively n = 3).
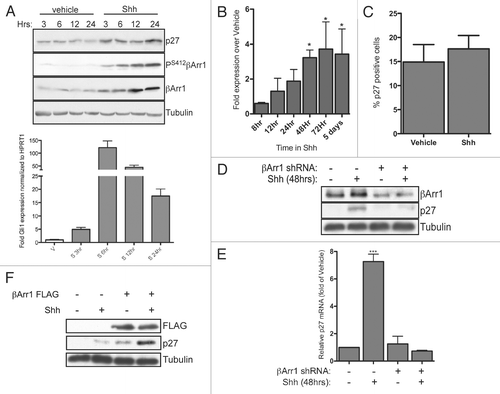
Figure 6 βArr1 is part of an immuno-complex with CREB, and both Shh and βArr1 regulate CREB and p300 binding to the p27 promoter. (A) CGNPs treated with vehicle or Shh for 48 h were collected for ChIP analysis with CREB antibody as described in the methods. Cells treated with cyclopamine were treated for 12 h prior to lysis. Analysis was performed by qPCR and results quantified as fold-over negative control primers. CREB is bound to the p27 but not the C-Jun promoter in response to Shh in CGNPs. (B) CGNPs were treated as above or infected with βArr1-targeting lentiviruses. Cells were collected for ChIP analysis using a p300 antibody and analysis performed by qPCR. p300 is recruited specifically to the p27 promoter in response to Shh, and this is reversed by βArr1 knockdown. (C) CGNP extracts were immunoprecipitated with IgG, CREB or PS412βArr1 antibody and the immunocomplexes were analyzed by western blots for IgG, CREB and PS412βArr1. 10% of the total lysate was loaded as a control. See also Supplemental Figure 4 for ChIP analysis with control antibodies.
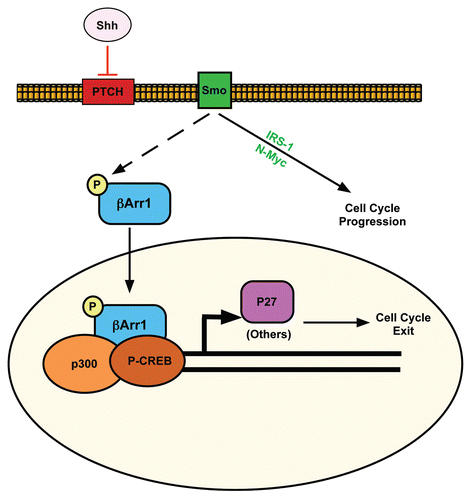
Figure 7 Proposed model for how Shh signaling pathway negatively regulates its mitogenic potential through β-arrestin 1. Activation of Shh signaling leads to cell cycle progression via the upregulation/activation of Gli, N-Myc and IRS1. Shh also activates a negative feedback loop through its induction and phosphorylation of βArr1, mediating its nuclear translocation, where it enhances p27 transcription in complex with CREB and p300. This sets the stage for accumulation of p27 to ultimately drive CGNP cell cycle exit.