Abstract
PLK1 is a critical mediator of G2/M cell cycle transition that is inactivated and depleted as part of the DNA damage-induced G2/M checkpoint. Here we show that down-regulation of PLK1 expression occurs through a transcriptional repression mechanism and that p53 is both necessary and sufficient to mediate this effect. Repression of PLK1 by p53 occurs independently of p21 and of arrest at G1/S where PLK1 levels are normally repressed in a cell cycle-dependent manner through a CDE/CHR element. Chromatin immunoprecipitation analysis indicates that p53 is present on the PLK1 promoter at two distinct sites termed p53RE1 and p53RE2. Recruitment of p53 to p53RE2, but not to p53RE1, is stimulated in response to DNA damage and/or p53 activation and is coincident with repression-associated changes in the chromatin. Down-regulation of PLK1 expression by p53 is relieved by the histone deacetylase inhibitor, trichostatin A, and involves recruitment of histone deacetylase to the vicinity of p53RE2, further supporting a transcriptional repression mechanism. Additionally, wild type, but not mutant, p53 represses expression of the PLK1 promoter when fused upstream of a reporter gene. Silencing of PLK1 expression by RNAi interferes with cell cycle progression consistent with a role in the p53-mediated checkpoint. These data establish PLK1 as a direct transcriptional target of p53, independently of p21, that is required for efficient G2/M arrest.
Introduction
The p53 tumor suppressor is a potent transcription factor that plays a pivotal role in preventing tumor development.Citation1–Citation4 p53 is induced in response to a variety of cellular stresses including DNA damage, leading mainly to the onset of apoptosis or cell cycle arrest at the G1/S and G2/M boundaries. In addition to activating the expression of a wide range of genes involved in these processes, p53 also represses the expression of many genes required for cell survival and cell cycle progression, including several involved in the G2/M transition.Citation5,Citation6 p53-mediated repression is still only partly understood and its ability to repress G2/M promoters may involve various mechanisms including interaction with the CCA AT-binding transcription factor NF-Y, direct binding to p53-responsive elements, and/or influence via the CDE/CHR (cell cycle-dependent element/cell cycle genes homology region).Citation7–Citation9
The polo-like kinases are a family of serine/threonine protein kinases that are structurally and functionally related to the prototypic Drosophila melanogaster family member, Polo.Citation10–Citation13 Mammalian PLK1 plays a pivotal role in the maturation of centrosomes, entry into M phase, spindle formation and cytokinesis.Citation10,Citation13 More recently, PLK1 was found to contribute to DNA synthesis where it plays a role in pre-replication complex formation.Citation14 PLK1 expression occurs at low levels during the early phase of the cell cycle and is mediated by a repression mechanism involving a promoter element termed CDE/CHR (cell cycle-dependent element/cell cycle gene homology region). However, its levels accumulate throughout S and G2 phase with a sharp increase in enzymatic activity occurring prior to the onset of M phase.Citation15–Citation18 PLK1 is oncogenic and constitutive expression of the enzyme causes transformation of NIH3T3 cells.Citation19 Additionally, PLK1 is upregulated in many human malignancies including colon cancer and is widely considered to be a potential therapeutic target.Citation13
p53 plays a critical role in the maintenance of the G2/M checkpointCitation6 and is protective against DNA damage-induced cell death.Citation20 PLK1 is phosphorylated and inactivated by ATM following DNA damage, leading to arrest at the G2/M boundary.Citation21 Similarly, DNA damage induces downregulation of PLK1 in an ATM/ATR-dependent fashion coincident with increases in p53 and p21.Citation22–Citation24 Several studies have suggested that this is dependent upon p53 and/or p21,Citation25–Citation28 but either have not provided any mechanistic insight or have attributed the downregulation to the CDE/CHR element,Citation28 a conclusion that could be explained by a p53-mediated G1/S arrest. Notably, PLK1 is required for recovery from DNA damage-induced G2 arrestCitation29 while constitutively active mutants of PLK1 can override the G2/M checkpoint.Citation23 These observations underscore the critical contribution of PLK1 activity towards this checkpoint and led us to examine the relationship between PLK1 and the p53 pathway in greater depth.
In the present study we confirm that PLK1 is downregulated following DNA damage. We show conclusively that p53 is both necessary and sufficient to mediate this effect and that it does so through direct repression of PLK1 expression. We find that p53 is present at two distinct sites in the PLK1 promoter and that its recruitment to one of these is further stimulated by DNA damage in a manner that is coincident with local changes in histone deacetylation favoring a closed chromatin structure. We also eliminate p21-mediated repression through the CDE/CHR element as a major factor in PLK1 repression. These data are consistent with the idea that PLK1 is quickly suppressed in a p53-dependent manner as a critical component of the G2/M checkpoint and have implications for PLK1 levels in tumor cells lacking functional p53.
Results
PLK1 is downregulated in a p53-dependent manner.
To determine whether PLK1 levels were regulated in response to DNA damage, three independent cell lines, MCF-7, OSA and U2OS cells (each of which have a wild type p53 response to DNA damage) were treated for 24 hours with the DNA methylating agent, cis-platin. As expected, the drug induced an increase in p53 levels in each cell type (); a corresponding increase in p53-Ser15 phosphorylation was observed in the MCF-7 cells that have significantly higher basal levels of wild type p53. Notably, in each case, the levels of PLK1 protein were decreased following the treatment with cis-platin (). Further analysis, by quantitative RT-PCR, indicated that a corresponding decrease in the level of PLK1 mRNA was observed () consistent with the idea the reduced levels of PLK1 protein reflect reduced levels of mRNA expression. In a control analysis, an increase in MDM2 expression was observed in all three lines confirming that the p53 response to DNA damage was intact in each of these lines. Treatment of the U2OS cells over a 24 h period with cis-platin or with the DNA strand break-inducer, etoposide, confirmed that PLK1 levels decrease coincidently with increased p53 levels and activation (Ser15 phosphorylation), and indicate that reduced PLK1 levels occur in response to more than one type of DNA damage-inducing drug ().
To determine whether the decrease in PLK1 levels was dependent upon the presence of p53, HCT116 cells (HCT116+/+) or an HCT116 derivative lacking expression of full-length p53 (HCT116−/−),Citation30 were treated with cis-platin. While a significant depletion of PLK1 levels was again noted in the p53-competent cells, no changes in PLK1 were evident in the p53-null cells (). These data indicate that cis-platin-induced PLK1 depletion is dependent upon wild type p53. Control analyses showed that increased expression of 14-3-3σ, MDM2 and p21 occurred only in the p53-competent cells confirming that the p53 pathway was functional only in the HCT116+/+ cells ().
p53 is necessary and sufficient to mediate decreased PLK1 levels.
To explore further the contribution of the p53 pathway towards downregulation of PLK1 HCT116 cells were treated with increasing amounts of Nutlin-3 which blocks the interaction of p53 with its binding site in the N-terminus of MDM2.Citation31 Following this treatment, a dose-dependent increase in p53 levels was accompanied by matching increases in the levels of p21, 14-3-3σ and MDM2 (). Notably, a dose dependent decrease in PLK1 levels was also observed. These changes were apparent only in the HCT116+/+ cells and not in the HCT116−/− cells, again consistent with a critical role for p53 in PLK1 reduction. Additionally, treatment of the HCT116+/+ cells, but not the HCT116−/− cells, with Nutlin-3 resulted in a dose-dependent decrease in PLK1 mRNA (). Furthermore, a time course analysis showed that the reduction in PLK1 levels was co-incident with the accumulation of p53 and p21 (). Strictly speaking, Nutlin-3 is an MDM2 inhibitor, raising the possibility that the changes in PLK1 levels could arise as a result of inhibition of a p53-independent function of MDM2. To address this issue, the HCT116−/− cells, in which endogenous MDM2 levels are barely detectable, were infected with adenoviruses expressing wild type MDM2 or, as a control, GFP. Treatment of the cells with Nutlin-3, in the absence or presence of MDM2, had no effect on PLK1 levels ruling out this trivial explanation (). (The MDM2 levels were slightly higher in the presence of Nultin-3 consistent with the idea that the drug may inhibit the ability of MDM2 to mediate its own destruction). While the data favor a mechanism whereby p53 can repress PLK1 expression, it is also possible that p53 may promote the turnover of PLK1 protein. However, analysis of the decay of PLK1 protein in the absence of Nutlin-3 indicated that its half life (determined by cycloheximide-chase) was not significantly different when compared with the decay rate following treatment of the cells with Nutlin-3 (compare and C). It is already established that PLK1 is degraded through the ubiquitin-proteasome pathway, most likely mediated by APC (reviewed in ref. Citation10).
PLK1 levels have been reported to be low during the early stages of the cell cycle and accumulate throughout S and G2 phases.Citation15–Citation18 These published observations were verified in the present study (Sup. Fig. 1). This effect is mediated through a repressor element, termed CDE/CHR, that operates during the early phase of the cell cycle.Citation18 The reduced level of PLK1 protein and -mRNA could therefore simply reflect the accumulation of cells at G1/S following a p53-dependent cell cycle arrest. To explore this possibility, SAOS-2 cells were used together with a derivative line (SAOS-2/p53,Citation32) in which wild type mouse p53 is inducibly expressed using a doxycycline-inducible promoter. (SAOS-2 cells, which lack endogenous p53, also lack Rb and cannot therefore arrest at G1/S). Upon treatment of the SAOS-2/p53 cells with doxycycline, p53 was induced in a dose-dependent () and time-dependent () manner leading to increases in the levels of p21 protein and mRNA; (p21 is both a p53 target and is primarily responsible for mediating p53-dependent G1/S arrest). Significantly, the levels of PLK1 protein ( and C) and mRNA () were depleted following induction of p53. These changes were not observed following doxycycline treatment of the parental SAOS-2 cells that lack the inducible p53. These data indicate that downregulation of PLK1 expression can be achieved in the absence of a p53-dependent G1/S arrest.
Using a parallel approach, the expression of p21 was silenced in two independent cell lines using siRNA (). Following treatment of the cells with Nutlin-3, significant downregulation of PLK1 levels were still observed even though there was no detectable p21 in the silenced cells. It should be noted, however, that the downregulation of PLK1 by Nutlin-3 was slightly less efficient in the p21-silenced cells as compared with the control treatments (especially in the HCT116 cells) suggesting that p21 is likely to make a minor contribution to this effect. To confirm this result, p21 expression was silenced in SAOS-2/p53 cells. Again, downregulation of PLK1 expression was independent of the presence of p21 (). PLK1 levels were also measured in HCT116+/+ cells null for p21Citation30 as compared with the HCT116+/+ and HCT116 p53−/− cells following induction of p53 (Sup. Fig. 2). The data indicate that downregulation of PLK1 levels by all three stresses (etoposide, Nutlin-3 and cis-platin) is partially impaired in the absence of p21, particularly at the time point measured (24 h post-treatment: see A). Nutlin-3, which is the most potent of these treatments in terms of inducing the p53 response, still retains significant ability to downregulate PLK1 levels. Notably, however, etoposide was also able to downregulate PLK1 levels in the p21-null cells at earlier time points although less robustly in comparison with the parental cells, and concomitantly with a much reduced induction of p53 [as measured by the increase in p53 levels and the induction of p53-responsive genes such as MDM2 and 14-3-3σ (Sup. Fig. 2A and C), and with a partial recovery by 24 h]. Flow cytometry confirmed that Nutlin-3 induces both a G1/S and G2/M block in the wild type cells but cell cycle arrest is abolished in the cells lacking p53 or p21 (Sup. Fig. 2B). This observation suggests that the p53-dependent decrease in PLK1 levels cannot simply be attributed to an arrest at G1/S where the PLK1 levels are naturally low.
p53 binds directly to the PLK1 promoter and inhibits PLK1 transcription.
The downregulation of PLK1 at both the protein and mRNA levels is consistent with the idea that p53 can mediate transcriptional repression of PLK1. If such a mechanism occurs then it should be possible to detect the presence and/or recruitment of p53 to the PLK1 promoter. To test this idea, chromatin immunoprecipitation (ChIP) experiments were carried out. Two potential p53 binding sites were observed in the PLK1 promoter at positions −2,067 to −2,016 [termed p53 responsive element 1 (p53RE1)] and −816 to −785 (p53RE2; ): the latter site contains two contiguous canonical p53 half-sites.Citation5 Accordingly, PCR primers were designed to permit analysis of each of these two regions. Additionally, given that p53 can repress promoters via interactions with factors operating within core promoter elements (for example through interaction with the CCAAT box-associated NF-Y transcription factorCitation7 or the CDE/CHR elementsCitation20) PCR primers were designed to permit analysis of the core promoter region. ChIP analysis of the PLK1 promoter revealed that p53 was present within both of the regions encompassing putative p53 consensus binding sites (p53RE1 and p53RE2 respectively: and C, left hand parts) but could not, however, be detected within the core promoter region containing the CAATT and CDE/CHR elements (). As control, no p53 was detected on the GAPDH promoter (). Interestingly, when p53 was induced either through treatment with etoposide or Nutlin-3 a modest increase in the levels of recruitment of p53 were observed at the region of the PLK1 promoter containing p53RE2, but not that containing p53RE1. This was accompanied by a striking decrease in the level of acetylated histone H3 present at p53RE2 (, right hand side), consistent with structural changes in the chromatin normally associated with transcriptional repression. Acetylated H3 was also observed in the vicinity of p53RE1 but was unaltered following stimulation of p53, indicating that the interaction of p53 with this region of the promoter did not mediate repression-associated chromatin remodeling.
To confirm that PLK1 expression is downregulated by a p53-dependent repression mechanism, HCT116 parental cells and the HCT116-p53−/− derivatives were treated with the histone deacetylase (HDAC) inhibitor, trichostatin A (TSA). Treatment of the cells with TSA alone led to a reduction in PLK1 levels independently of p53 (): this observation probably reflects the widespread involvement of HDACs in regulating gene expression; (its p53-independent effect on PLK1 expression is most plausibly explained if TSA relieves expression of a PLK1 negative regulator). However, treatment of the parental cells, but not the p53−/− cells, with Nutlin-3 alone led to a significant downregulation of PLK1 levels over and above that seen with TSA alone. This inhibition was relieved in the presence of TSA where the PLK1 levels recovered to the level of PLK1 in the cells that had been treated with TSA alone. These data support the idea that p53-mediated downregulation of PLK1 expression occurs via HDAC-mediated transcriptional repression. To provide a more focussed examination of the involvement of histone deacetylase, quantitative ChIP analysis was carried out using an antibody that recognizes HDAC1 (). The data indicate that the presence of HDAC1 is enriched within the vicinity of p53RE2 in response to etoposide treatment consistent with the idea that HDAC1 is recruited to the promoter as part of the repression mechanism. In contrast, the presence of HDAC1 on the p53-stimulated p21 promoter was reduced under these conditions, as might be expected with activation of this promoter ().
To determine whether p53 can bind specifically to the PLK1 promoter, EMSA experiments were carried out using complementary DNA oligonucleotides representing p53RE2 [: p53RE2 was examined because it is responsive to stimuli that activate p53 (see above) and is located in the vicinity of the repression-associated histone deacetylation events]. Bacterially-expressed p53 bound to the p53RE2 promoter element and its binding was both shifted and stimulated by the anti-p53 C-terminal antibody, PAb421. [The p53 protein has both a site-specific binding function (encompassed within the core domain of the protein) and a non-specific DNA binding function (located at the C-terminus): the PAb421 antibody is an established activator of the site-specific-, but not the non-specific-DNA binding function of p53].Citation33 p53 was still able to interact with a mutant of the p53RE2 response element in which three base pairs were changed. However, this binding, although super-shifted by PAb421, was not stimulated by the antibody suggesting that it occurred through the non-specific binding function of p53. By comparison, similar differences in site-specific and non-specific binding are illustrated in the controls for p53/DNA binding where the appropriate sequences used by Hupp and colleagues were employed.Citation33 These data support the idea that p53RE2 constitutes an effective p53 binding site.
p53 represses the expression of the PLK1 promoter in a promoter-reporter assay.
To determine whether p53 can directly repress expression from the PLK1 promoter, 2.4 kb of the promoter was cloned upstream of a luciferase reporter gene (). The promoter-luciferase plasmid was transfected into SAOS-2 cells () or HCT116-p53−/− cells (Sup. Fig. 3) together with increasing amounts of a plasmid expressing wild type human p53. These analyses showed that very low levels of p53 (low nanogram amounts of plasmid) inhibit expression [; similar levels of p53 are required to stimulate the expression from the p21 promoter (data not shown)]. Interestingly, repression mediated by increasing amounts of p53 reached a threshhold (with generally 20–40% of the activity remaining) beyond which further increases in the amount of p53 had no further effect. Two tumor-associated inactive mutants of p53 that cannot bind DNA failed to repress the PLK1 promoter ( and C). Additionally the L22Q/W23S mutant of p53 (), which is inactive in transactivation,Citation34 was also unable to repress the PLK1 promoter suggesting that this domain participates in the repression mechanism. (Others have established previously that this region of p53 also contributes to transcriptional repression).Citation35 Mutation of the CDE/CHR element did not eliminate repression by p53 () indicating that p53-mediated repression occurs independently of this important repressor element: however, the basal levels of expression from the PLK1 promoter increased by 2–3-fold consistent with published data indicating that this element mediates cell cycle-dependent repression of PLK1.Citation18 Titration of a plasmid encoding active p21 [which was able to arrest cells at G1/S (data not shown)] had no significant effect on expression from the PLK1 promoter although the trend over several experiments suggested the possibility of a small contribution by p21 ().
To determine whether the repression was mediated through p53RE2, the same three mutations that were present in the mutant p53RE2 element used in the EMSA experiments () were introduced into the PLK1 promoter-luciferase plasmid. PLK1 promoter-luciferase analysis with this mutant promoter indicated that the p53 responsiveness does indeed appear to be diminished (). Additionally, however, there was a striking reduction in the basal level of expression suggesting that one or more of the bases we had changed are vital to maintaining basal PLK1 expression (). This observation suggests the possibility that the binding site for a stimulatory factor may overlap with the p53 binding site.
PLK1 depletion causes a mitotic arrest leading to p53-independent apoptosis.
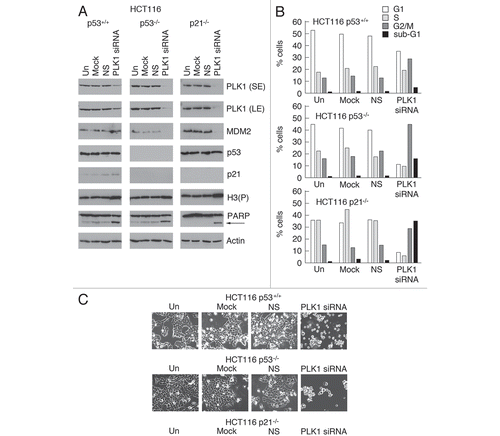
PLK1 plays an essential role in the transition of the cell cycle from late G2 phase, throughout mitosis to cytokinesisCitation10 and its downregulation has been proposed to contribute to the G2/M checkpoint.Citation22–Citation24 To determine the outcome of downregulating PLK1 levels on cell cycle progression PLK1 expression was silenced by siRNA. Knock-down of PLK1 in the HCT116 (p53+/+) cells () led to cell cycle arrest at both the G1 and G2/M phases of the cycle with a small proportion of the cells undergoing apoptosis as evidenced by flow cytometry (), and PARP cleavage (). The presence of increased levels of phosphorylated histone H3 in these cells, and their rounded-up appearance (), suggested that they had entered mitosis before undergoing p53-independent apoptosis. When p53 or p21 were absent there was no G1/S arrest with the outcome that there was a higher proportion of cells at G2/M and a significantly increased level of apoptosis. These data suggest that p53 may play an additional role by protecting the cells from loss of PLK1, presumably by holding a proportion of the cells at the G1/S boundary. The finding that loss of PLK1 permits the cells to enter M phase but terminate in apoptosis is consistent with the idea that p53 may mediate a checkpoint that leads to cell death through the downregulation of PLK1. Alternatively, however, it is possible that a G2/M cell cycle arrest may indeed involve p53-mediated depletion of PLK1, but may additionally require other p53-coordinated events to hold the cells at the G2/M boundary thereby halting any commitment towards undergoing cell death.
Discussion
In the present study we show that PLK1 protein and -mRNA levels are depleted in response to DNA damage-inducing agents in a p53-dependent manner. Moreover, the use of Nutlin-3, which inhibits the p53-MDM2 interaction,Citation31 or expression of p53 via an inducible system demonstrated that induction of p53 alone is sufficient to achieve this effect in the absence of DNA damage. These data suggest that PLK1 is likely to be a target of p53 not only following DNA damage but also in response to other stimuli that induce a p53 response. The data also suggest that p53 can mediate these effects by directly repressing expression of PLK1. In support of this idea we find that p53 is recruited to two distinct regions within the PLK1 promoter. In one case (p53RE1) this is associated with an open chromatin structure that is unaffected by p53 stimulation. In the other case however (p53RE2) the chromatin conformation becomes closed (as evidenced by loss of histone acetylation) in response to p53 activation and increased p53 recruitment (). The involvement of histone deacetylase in the p53-dependent downregulation of PLK1 additionally supports a repression model (). At present we do not know what additional components are required for this repression mechanism, nor do we know the sequence of events involved or the means by which the repression is regulated. We have, however, been able to rule out certain mechanisms. For example, p21-dependent or -independent repression via the CDE/CHR element, while relevant to some promoters (reviewed in ref. Citation20), and which has previously been suggested by others to mediate the ability of p53 to regulate PLK1 levels,Citation28 does not appear to be a major contributor to repression of PLK1 (, and and Sup. Fig. 2). The finding that p53 could not be detected close to the transcriptional start site () also suggests that the inhibition of DNA-bound NF-Y that occurs in promoters such as the CDC2 and Cyclin B2 promoters is unlikely to mediate repression of PLK1.Citation7
The finding that mutation of the p53 responsive element, p53RE2, leads to a loss in basal PLK1 expression suggests that a transcription factor that is crucial for maintaining basal PLK1 levels binds to a site that overlaps with p53RE2. It also raises the possibility that part of the repression mechanism may involve “competitive displacement” by p53, a mechanism that is known to mediate repression of several other p53-responsive promoters,Citation20,Citation36,Citation37 in some cases (e.g., CDC25CCitation20) as part of a dual repression mechanism. Initial analysis of the p53RE2 sequence suggests a list of at least 20 different transcription factors that could bind to this region. Identifying this factor will therefore be an important goal in exploring the elements of this repression mechanism in depth.
Our data also demonstrate that PLK1 satisfies the five conditions that normally constitute a p53-responsive gene.Citation5 These are: (1) presence of a demonstrable p53-responsive element in the promoter ( and ); (2) downregulation at the RNA and protein level by wild type, but not mutant, p53 (– and ); (3) p53 regulation conferred on a test gene through the responsive element (); (4) the presence of p53 on the endogenous promoter within the vicinity of the response element as revealed by ChIP (); and (5) sequence-selective binding of p53 to the RE in vitro (). Our data support and significantly extend previous observations suggesting that PLK1 may be downregulated by a p53-dependent repression mechanism.Citation21–Citation24,Citation26,Citation27
Several laboratories have examined the outcome of silencing expression of PLK1 and broadly agree that depletion of PLK1 leads to several defects associated with the transition through mitosis and cytokinesis, resulting in mitotic catastrophe and/or apoptosis.Citation14,Citation38–Citation44 Interestingly, PLK1 depletion has also been reported to lead to DNA damage.Citation14,Citation41 Since p53 mediates the repression of PLK1 [both at the levels of expression and posttranslational modification (this study: reviewed in ref. Citation22–Citation27)], we re-examined the effects of depleting PLK1 to determine the likely consequence of its downregulation following p53 induction. Consistent with published studies, we find that PLK1 knock-down arrests the cells not in G2 but in mitosis, as judged by flow cytometry profile, cellular morphology and the presence of phosphorylated histone-H3 (a marker for mitotic cells; ). Notably, these cells fail to complete mitosis [as evidenced by the reduction in the G1 peak (in the p53-null and p21-null derivatives)—i.e., they are not re-entering cycle] and undergo apoptosis, as shown by the presence of a sub-G1 population of cells together with the appearance of cleaved PARP. These observations raise some curious questions about the impact of inducing p53 on late cell cycle events. On the one hand, the downregulation of PLK1 by p53 could lead indirectly to apoptosis by allowing cells to progress towards mitosis when they lack this critical mitosis orchestrator. Arguing against this, however, is the observation that downregulation of PLK1 does not lead to any significant increase in apoptosis when it is achieved through inducing p53 (as judged by the lack of any increase in the sub-G1 population—see Sup. Fig. 2). A more plausible explanation is that p53 stably maintains arrest in G2 through a series of events including downregulation of CDC25C, Cyclin B1, CDC2, Stathmin, MAP4, etc., (reviewed in ref. Citation6 and Citation45) that are by necessity coordinated and inter-dependent, and that it is within this context that the inhibition of PLK1 functions as an important and integral element in this orchestration. We also note that arrest in mitosis followed by apoptosis occurs not only in the HCT116 parental cells but also in the p53-null and p21-null derivatives (Sup. Fig. 2). Strikingly, however, the level of apoptosis is significantly greater in the absence of p53, and even greater still when p21 is absent. A protective effect of p53, consistent with these observations, has also been noted by others.Citation14,Citation27,Citation38,Citation41,Citation42 We suggest, from the flow cytometry profiles in Supplemental Figure 2B, that the ability of p53 and/or p21 to arrest the cells in G1 may afford this protection, at least in part, by preventing the cells from progressing to the G2/M stages of the cell cycle where the absence of PLK1 becomes a critical problem. Additionally, however, the recent demonstration that PLK1 is required for pre-replication complex formation during DNA synthesis, and consequently that depletion of PLK1 leads to DNA damage,Citation14,Citation41 suggests that p53 may be alerted to abnormally low PLK1 levels through the DNA damage pathways and provides additional protection by blocking entry into and/or progression through S phase.
Our data are also consistent with the growing awareness that p53 status may have a profound influence on PLK1 levels in tumors.Citation27 Our findings thus provide mechanistic insight into how mutation of p53 or loss of p53 function, during tumorigenesis can lead to, or contribute towards, unregulated PLK1 levels and, consequently, to an augmented hyper-proliferative drive. They also support the idea, expressed by other researchers,Citation27,Citation38,Citation42 that an intact p53 pathway may assist the selective treatment of tumor cells with PLK1 inhibitors by affording a level of protection to normal cells.
Materials and Methods
Cell culture.
Cells were maintained in McCoy's 5a medium [Biosera; HCT116 (p53+/+, p53−/− and p21−/−) colon carcinoma cellsCitation30] were maintained in McCOY's 5a medium (Biosera) or Dulbecco's modified Eagle's medium (Biosera; OSA, U2OS, MCF-7 and SAOS-2), supplemented with 10% heat-inactivated fetal bovine serum (Biosera), penicillin (100 IU/ml)/streptomycin (100 µg/ml) (Invitrogen) and 2 mM Glutamine (Invitrogen). Cultures were maintained at 37°C in a humidified 5% CO2 atmosphere.
Flow cytometry.
Cells were treated with 5 µM Nutlin-3 or untreated as control, for 24 h. Cells were analyzed by flow cytometry following labeling with BrdU and propidium iodide as described previously.Citation32
Luciferase reporter assays.
A PLK1-luciferase reporter plasmid was constructed by cloning 2.3 kb of the human PLK1 promoter (base numbers 1,590–3,893 from the Genbank sequence X90725) upstream of the firefly luciferase gene in the plasmid pGL3-basic (Promega). Cells (3 × 104/well) were seeded onto a 24-well plate and transfected in triplicate using Fugene (Roche) as indicated in the figure legends. Transfected cells were harvested 24 hr post-transfection by adding 100 µl of passive lysis buffer (Promega) per well. 20 µl of protein extract was analyzed in a luminometer using the Dual Luciferase Reporter Assay Kit (Promega). Variations in transfection efficiencies were corrected by determining the Renilla luciferase activity of the sample.
Site-directed mutagenesis.
Point mutations at the CDE/CHR site in the PLK1 promoter-luciferase plasmid were generated using the Stratagene Quik-Change system according to the manufacturer's instructions. The complementary mutant oligonucleotides were as follows with the mutant bases shown underlined:
Coding strand oligonucleotide: 5′-GTA ACG TTC CCA TAT CCG CGT TCA CAT TCG GGG AGG AG-3′
Non-coding strand oligonucleotide: 5′-CTC CTC CCC GAA TGT GAA CGC GGA TAT GGG AAC GTT AC-3′.
Gene silencing using siRNA.
HCT116 and U2OS cells were transfected with PLK1 or p21 siRNA (ON-TARGETplus SMARTpool L-003290-00 or L-003471-00 respectively Dharmacon) using Lipofectamine (for U2OS cells) or by reverse transfection using Lipofectamine RNAiMAX Transfection Reagent (for HCT116 cells) in a 6-well plate according to the manufacturers' instructions.
Real-time (Taqman) RT-PCR.
Total RNA was isolated from cells using the RNeasy Mini Protocol according to the manufacturer's instructions (Qiagen Ltd.) cDNA was synthesized using standard reverse transcription PCR techniques. Steady-state levels of mRNA encoding PLK1 and MDM2, were determined by real-time quantitative PCR as described previously.Citation46 The primers and probe for PLK1 amplification were as follows.
PLK1 Forward primer: 5′-GGA TCA CAC CAA GCT CAT CTT G-3′
PLK1 Reverse primer: 5′-CCC GCT TCT CGT CGA TGT-3′
PLK1 Probe: 5′-CCC ACT GAT GGC AGC CGT GAC C-3′
The primers and probes for MDM2 were as follows.
MDM2 Forward primer: 5′-CAA GTT ACT GTG TAT CAG GCA GGG-3′
MDM2 Reverse primer: 5′-CCT TCC ATC ACA TTG CAA CAG A-3′
MDM2 Probe: 5′-TTG GAA ATG CAC TTC ATG CAA TGA AAT GAA TCC-3′
Antibodies and western blot analysis.
SDS-PAGE and western blotting was carried out using standard conditions. Nitrocellulose membranes were probed for the presence of PLK1 (antibodies PL2 and PL6, Zymed Laboratories Inc.), p53 (antibody DO1, Moravian Biotechnology; or CM-1Citation47), p53 phospho-Ser15 (Ab-3, Calbiochem), MDM2 (SMP14, D12, Santacruz Biotechnology, Inc.; or 4B2, Moravian Biotechnology), p21 (H-164, Santacruz Biotechnology, Inc.), 14-3-3σ (sc-7683, Santacruz Biotechnology, Inc.), phospho-Histone H3 (06-570, Upstate Biotechnology), acetyl-lysine (4G12, Upstate Biotechnology), HDAC1 (sc-6298, Santacruz Biotechnology, Inc.) or actin (20-33, Sigma-Aldrich). Secondary antibodies used were HRP-conjugated rabbit antimouse (DAKO), -goat anti-rabbit (DAKO) or -donkey anti-goat (sc-2020, Santacruz Biotechnology, Inc.). Proteins were detected by enhanced chemiluminescence according to the manufacturer's instructions (Pierce Biotechnology, Inc.).
Cell lysis.
Cells were lysed in 50 mM Tris-HCl, pH 7.5, 1% NP-40, 0.25% Sodium deoxycholate, 150 mM NaCl, 5 mM EDTA, 50 mM Sodium fluoride, 1 mM Sodium orthovanadate, 1 mM Benzamidine and a protease inhibitor cocktail (Calbiochem), left on ice for 10 mins, followed by centrifugation at 13,000 rpm for 10 mins at 4 degrees. The soluble fractions were collected.
Infection with recombinant adenovirus.
Recombinant viruses Ad-MDM2, Ad-GFP and Ad-CON have been described elsewhere.Citation48 HCT116−/− cells were grown to 80% confluency and infected at an MOI of 50, followed by an incubation period at 37°C for 4 h (35). Cells were then treated with 10 µM Nutlin-3,Citation31 and incubated at 37°C for a further 24 h. Extracts were analyzed by western blotting.
Quantitative chromatin immunoprecipitation (ChIP).
The promoter sequence of human PLK1 (accession number X90725) was screened for potential p53 binding sites using in-house software. This approach identified potential p53 binding sites at positions, relative to the transcriptional start site, −2,086 to −2,016 [p53 binding site-1 (p53RE-1)] and −816 to −785 [p53 binding site-2 (p53RE2)]. The sequence of these regions is given in . Primers and probes for quantitative chromatin immunoprecipitation analysis were designed using Primer Express software (Applied Biosystems) to allow amplification of the regions encompassing the two potential p53 binding sites and, additionally, the CAAT and CDE/CHR signals (both of which are located close to the transcriptional start site). The primers and probes were as follows:
p53 binding site-1 Forward primer: 5′-CAC TGT GCC TAA ACT GTT CGT ACA-3′
p53 binding site-1 Reverse primer: 5′-CAC ATA TGA AAA TCT GAC TCC CAG AA-3′
p53 binding site-1 Probe: 5′-TGT GGG TTA TGC CCA ACA TCT GCT TTC-3′
p53 binding site-2 Forward primer: 5′-TGC TAG GCA TTC TTC CAA CCT T-3′
p53 binding site-2 Reverse primer: 5′-GAA TTT CCC AGG GCC TAA AAT AG-3′
p53 binding site-2 Probe: 5′-CCT CCC TCT GAC CAA GAA ACT GAG TGT CC-3′
CAATT box Forward primer: 5′-ATC CCC GCG GCC AAT-3′
CAATT box Reverse primer: 5′-TCC TCC CCG AAT TCA AAC G-3′
CAATT box Probe: 5′-CGC GCA GGC TTT TGT AAC GTT CCC-3′
p21 promoter, distal p53 binding site Forward primer: 5′-GTG GCT CTG ATT GGC TTT CTG-3′
p21 promoter, distal p53 binding site Reverse primer: 5′-CTG AAA ACA GGC AGC CCA AG-3′
p21 promoter, distal p53 binding site, probe: 5′-TGG CAT AGA AGA GGC TGG TGG CTA TTT TG-3′
For the quantitative ChIP analysis, U20S or HCT116 cells were treated with up to 50 µM Etoposide, 20 µM Cis-platin or 10 µM Nutlin-3 for 6 or 8 h and chromatin immunoprecipitation was performed as described previously.Citation32,Citation49
Abbreviations
| ChIP | = | chromatin immunoprecipitation |
| PLK1 | = | polo-like kinase-1 |
| p53RE1 and p53RE2 | = | p53 responsive elements-1 and -2 respectively |
Figures and Tables
Figure 1 DNA damage downregulates PLK1 protein and mRNA levels in a p53-dependent manner. (A) MCF-7, OSA and U2-OS cells (all expressing wild type p53) were treated with 20 µM Cis-platin for 24 h. Cells were lysed and western blotting performed with antibodies as indicated. (B) MCF-7, OSA and U2-OS cells were treated as in (A). RNA was extracted and steady-state levels of mRNA encoding PLK1 and MDM2 were determined by real-time quantitative PCR. (C) U2OS cells were treated with 20 µM cis-platin or -etoposide. Cells were harvested at the indicated times and examined by western blotting. (D) HCT116 p53+/+ or p53−/− cells were treated with 20 µM Cis-platin for 24 h. Cells were lysed and western blotting performed with antibodies as indicated.
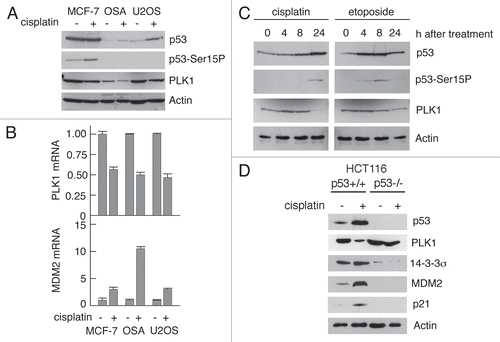
Figure 2 Nutlin-3 induces p53-mediated downregulation of PLK1. (A) HCT116 p53+/+ or p53−/− cells were treated with 0–10 µM Nutlin-3 or DMSO as vehicle control. The cells were lysed and western blotting was performed with the indicated antibodies. (B) HCT116 cells were treated as in (A) and the steady-state levels of mRNA encoding PLK1 and MDM2 were determined by real-time quantitative PCR. (C) HCT116 p53+/+ cells were treated with 5 µM Nutlin-3 for 0–24 h. Cells were lysed and western blotting performed with the antibodies as indicated. (D) HCT116 p53−/− cells were infected with recombinant viruses Ad-MDM2 or Ad-GFP at an MOI of 50 for 4 h. The cells were then treated with 10 µM Nutlin-3 for 24 h and lysed before western blotting was performed with the indicated antibodies. (E) HCT116 p53 wild-type cells were treated with 10 µg/ml Cycloheximide and cell lysates prepared at the indicated time points after addition. Western blotting was then performed with the antibodies as indicated.
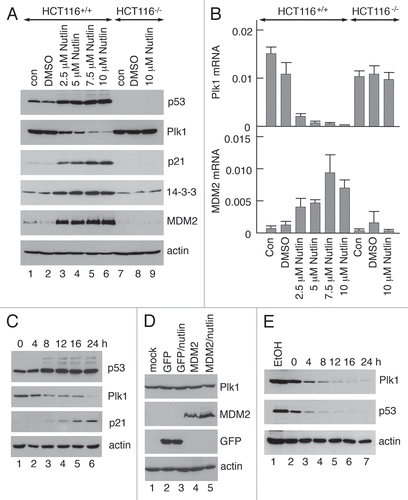
Figure 3 Downregulation of PLK1 levels in SAOS-2 cells following inducible ectopic expression of p53. SAOS-2 cells and a derivative line that ectopically expresses mouse p53 in a doxycycline-inducible mannerCitation32 were treated for 24 h in the presence of increasing amounts of doxycycline. After harvesting, the cell extracts were analysed by western blotting (A) or quantitative RT-PCR (B) to determine the levels of PLK1, p53, p21 and, as control, actin. The cells were also treated with 0.5 mM doxycycline for up to 34 h prior to harvesting. The levels of PLK1, p53, p21 and actin were subsequently determined by western blotting (C).
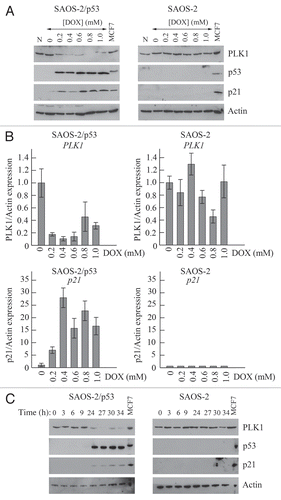
Figure 4 Downregulation of PLK1 can occur following elimination of p21. (A) Expression of p21 in HCT116 cells or U2OS cells was silenced by siRNA. Control cells were untreated (Unt), mock-transfected (Mock), or transfected with a non-silencing siRNA. Cells were subsequently treated with 5 µM Nutlin-3 for 24 h prior to harvesting. Western blotting was subsequently performed with the antibodies as indicated. (B) Expression of p21 in SAOS-2 cells expressing p53 under an IPTG-inducible system (or parental cells lacking p53, as control) was silenced by siRNA treatment as in (A). Western blotting was subsequently performed with the antibodies as indicated.
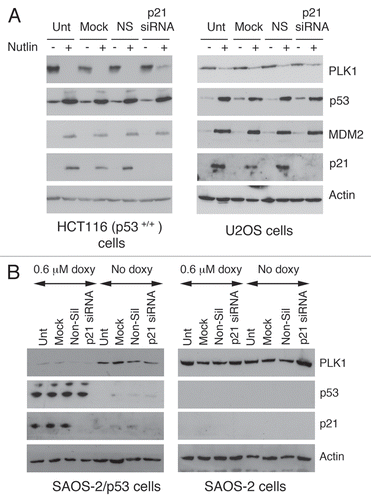
Figure 5 p53 is recruited to the PLK1 promoter in response to DNA damage. (A) Schematic showing the PLK1 promoter. The positions of the two putative p53 responsive elements (p53RE1 and p53RE2) are shown together with the CAATT box and the CDE/CHR element. The numbers in parentheses indicate the positions of these elements relative to the transcriptional start site. The arrows show the positions of primers/probes designed to examine these regions by quantitative ChIP. The sequences of the regions containing potential p53 responsive elements p53RE1 and p53RE2 are shown beneath and the bases within these elements are shown in upper case. Quarter sites are indicated by arrows. The sequence information is taken from Genbank sequence X90725. (B–E) HCT116 cells were treated with 50 µM etoposide (Etop), 10 µM Nutlin-3 (Nutl) or were untreated (Un) as control. Cells were harvested after 8 h and the presence of p53 and acetylated histone H3 on the p53RE1 (B) and p53RE2 (C) regions of the PLK1 promoter were determined by ChIP. Control immunoprecipitations were carried out using IgG. ChIP analyses was extended to the core promoter region where the CAATT box and CDE/CHR elements are located (D). As a negative control, the GAPDH promoter was examined for the presence of p53 (E). The measurements were carried out in triplicate and the results are representative of three independent experiments.
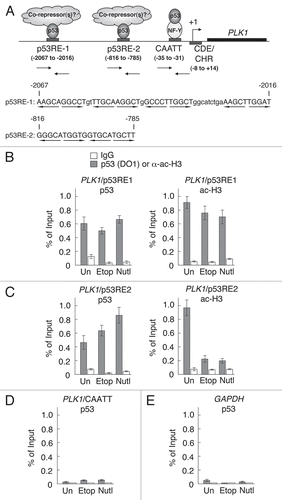
Figure 6 Histone deacetylase activity contributes to downregulation of PLK1 by p53. (A) HCT116 parental cells or the p53-null derivative were treated with 1 µM trichostatin A (TSA) alone, 10 µM Nutlin-3, or a combination of these two drugs. Cells were lysed after 24 h and western blotting performed with antibodies as indicated. (B) HCT116 cells were treated for 8 h with 50 µM etopoide. Chromatin immunoprecipitation analysis was carried out using an antibody that recognizes HDAC1.
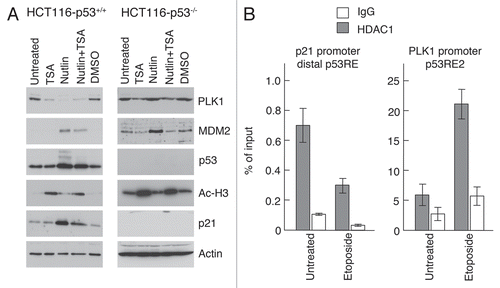
Figure 7 p53 binds directly to the p53RE2 sequence. (A) EMSA was carried out using radiolabeled dsDNA probes corresponding to the p53RE2 sequence (lanes 1–3) and a mutant derivative (lanes 4–6) in which three base pairs were substituted to destroy the p53 binding site consensus: [the sequence GTG CAT GCT T was changed to GTG gAT tgT T (with the changed bases shown in lower case)]. (B) EMSA showing the binding of p53 to the control sequences PG (comprising a canonical p53 responsive element, lanes 1–3) and TL (comprising a sequence unrelated to a canonical p53 binding site, lanes 4–6). These reagents are established tools with which to measure site-specific p53 binding.Citation33
![Figure 7 p53 binds directly to the p53RE2 sequence. (A) EMSA was carried out using radiolabeled dsDNA probes corresponding to the p53RE2 sequence (lanes 1–3) and a mutant derivative (lanes 4–6) in which three base pairs were substituted to destroy the p53 binding site consensus: [the sequence GTG CAT GCT T was changed to GTG gAT tgT T (with the changed bases shown in lower case)]. (B) EMSA showing the binding of p53 to the control sequences PG (comprising a canonical p53 responsive element, lanes 1–3) and TL (comprising a sequence unrelated to a canonical p53 binding site, lanes 4–6). These reagents are established tools with which to measure site-specific p53 binding.Citation33](/cms/asset/56a9766a-fc9c-439f-a186-0940da4f0e12/kccy_a_10913532_f0007.gif)
Figure 8 p53 represses expression of the PLK1 promoter. SAOS-2 cells were transfected in triplicate with increasing amounts a plasmids encoding wild-type or mutant p53 or p21, together with promoter/(Firefly) luciferase vectors containing a wild type or mutated PLK1 promoter and, as an internal standard, a plasmid expressing Renilla luciferase. Cell extracts were made 24 h after transfection and the levels of luciferase were determined. The graphs show the ratios of firefly luciferase/Renilla luciferase with error bars representing the standard deviations. Western blots shown under each graph show representative increases in the levels of expression of p53 or p21 with actin as loading control.
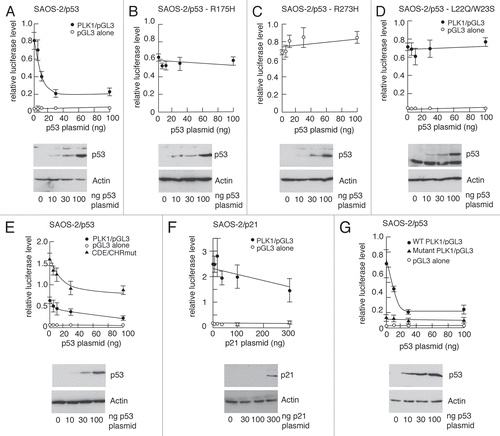
Figure 9 Silencing of PLK1 expression leads blocks cell cycle progression and initiates apoptosis. PLK1 expression in parental HCT116 cells, and in the p53-null and p21-null derivatives, was silenced by siRNA transfection. The cells were harvested 24 h after transfection. (A) western blotting was performed with the antibodies as indicated. (B) Cell were analysed by Flow cytometry. A set of data is shown that is representative of three independent experiments. (C) The cells were examined by phase contrast microscopy.
Additional material
Download Zip (4.8 MB)Acknowledgements
This work was supported by grants from Cancer Research UK and the Association for International Research.
References
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749 - 758
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev 2007; 8:275 - 283
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413 - 431
- Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol 2009; 1:a001883
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402 - 412
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20:1803 - 1815
- Imbriano C, Gurtner A, Cocchiarella F, Di Agostino S, Basile V, Gostissa M, et al. Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol 2005; 25:3737 - 3751
- Martin BT, Strebhardt K. Polo-like kinase 1: target and regulator of transcriptional control. Cell Cycle 2006; 5:2881 - 2882
- Saint Clair S, Manfredi JJ. The dual specificity phosphatase Cdc25C is a direct target for transcriptional repression by the tumor suppressor p53. Cell Cycle 2006; 5:709 - 713
- Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol 2009; 10:265 - 275
- Fenton B, Glover DM. A conserved mitotic kinase active at late anaphase-telophase in syncytial Drosophila embryos. Nature 1993; 363:637 - 640
- Glover DM, Hagan IM, Tavares AA. Polo-like kinases: a team that plays throughout mitosis. Genes Dev 1998; 12:3777 - 3787
- Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006; 6:321 - 330
- Yim H, Erikson RL. Polo-like kinase 1 depletion induces DNA damage in early S prior to caspase activation. Mol Cell Biol 2009; 29:2609 - 2621
- Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci 1994; 107:1509 - 1517
- Hamanaka R, Smith MR, O'Connor PM, Maloid S, Mihalic K, Spivak JL, et al. Polo-like kinase is a cell cycle-regulated kinase activated during mitosis. J Biol Chem 1995; 270:21086 - 21091
- Lee KS, Yuan YL, Kuriyama R, Erikson RL. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol Cell Biol 1995; 15:7143 - 7151
- Uchiumi T, Longo DL, Ferris DK. Cell cycle regulation of the human polo-like kinase (PLK) promoter. J Biol Chem 1997; 272:9166 - 9174
- Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, et al. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun 1997; 234:397 - 405
- St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, et al. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell 2004; 16:725 - 736
- van Vugt MA, Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005; 24:2844 - 2859
- Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem 2004; 279:25549 - 25561
- Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase 1 is a target of the DNA damage checkpoint. Nat Cell Biol 2000; 2:672 - 676
- van Vugt MA, Smits VA, Klompmaker R, Medema RH. Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J Biol Chem 2001; 276:41656 - 41660
- Incassati A, Patel D, McCance DJ. Induction of tetraploidy through loss of p53 and upregulation of Plk1 by human papillomavirus type-16 E6. Oncogene 2006; 25:2444 - 2451
- Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, et al. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem 2004; 279:21183 - 21192
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr, Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA 2009; 106:3964 - 3969
- Zhu H, Chang BD, Uchiumi T, Roninson IB. Identification of promoter elements responsible for transcriptional inhibition of polo-like kinase 1 and topoisomerase IIalpha genes by p21(WAF1/CIP1/SDI1). Cell Cycle 2002; 1:59 - 66
- van Vugt MATM, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell 2004; 15:799 - 811
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 1501
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 848
- Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J, et al. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J 2005; 24:543 - 553
- Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell 1992; 71:875 - 886
- Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55 kD protein. Genes Dev 1994; 8:1235 - 1246
- Hammond EM, Mandell DJ, Salim A, Krieg AJ, Johnson TM, Shirazi HA, et al. Genome-wide analysis of p53 under hypoxic conditions. Mol Cell Biol 2006; 26:3492 - 3504
- Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 2002; 277:3247 - 3257
- Lee KC, Crowe AJ, Barton MC. p53-mediated repression of alpha-fetoprotein gene expression by specific DNA binding. Mol Cell Biol 1999; 19:1279 - 1288
- Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y. Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res 2005; 65:2698 - 2704
- Kreis NN, Sommer K, Sanhaji M, Kramer A, Matthess Y, Kaufmann M, et al. Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1). Cell Cycle 2009; 8:460 - 472
- Liu X, Erikson RL. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc Natl Acad Sci USA 2002; 99:8672 - 8676
- Liu X, Erikson RL. Polo-like kinase (PLK)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA 2003; 100:5789 - 5794
- Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 2006; 26:2093 - 2108
- Spankuch-Schmitt B, Bereiter-Hahn J, Kaufmann M, Strebhardt K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst 2002; 94:1863 - 1877
- Spankuch-Schmitt B, Wolf G, Solbach C, Loibl S, Knecht R, Stegmuller M, et al. Downregulation of human polo-like kinase activity by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene 2002; 21:3162 - 3171
- Stark GR, Taylor WR. Control of the G2/M transition. Mol Biotechnol 2006; 32:227 - 248
- Young V, Ho M, Vosper H, Belch JJ, Palmer CN. Elevated expression of the genes encoding TNFalpha and thromboxane synthase in leucocytes from patients with systemic sclerosis. Rheumatology 2002; 41:869 - 875
- Midgley CA, Fisher CJ, Bartek J, Vojtesek B, Lane D, Barnes DM. Analysis of p53 expression in human tumours: an antibody raised against human p53 expressed in Escherichia coli. J Cell Sci 1992; 101:183 - 189
- Kowalik TF, DeGregori J, Leone G, Jakoi L, Nevins JR. E2F1-specific induction of apoptosis and p53 accumulation, which is blocked by Mdm2. Cell Growth Differ 1998; 9:113 - 118
- Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NFkappaB subunit. EMBO J 2006; 25:4820 - 4832