Abstract
Lung cancer remains the foremost cause of cancer deaths worldwide. Despite advances in both detection and treatment, diagnosis is often late and the prognosis for patients poor. Our understanding of the molecular basis and progression of lung cancer remains incomplete, hampering the design and development of more effective diagnostic tools and therapies for this devastating disease. However, the last twelve months have witnessed the publication of several studies that represent significant advances in our knowledge of lung cancer, and may represent important steps on the road to effective new therapies. In this review we aim to summarize these recent developments, and give our perspectives on the therapeutic possibilities they may offer in the future.
Introduction
Since 1985 lung cancer has been both the most prevalent and most lethal cancer in the world.Citation1 In 2002 there were more than 1.15 million recorded deaths from lung cancer, with 1.35 million new cases reported, representing around one eighth of all cancer diagnoses.Citation2 It is by far the most commonly diagnosed cancer in men, although the incidence rate in 2002 was around three times lower in women. Since 1980 the proportion of lung cancer patients living in the developing world has risen sharply, from 31 to 50% of total cases worldwide.
Lung cancer itself is defined as a malignancy arising from the cells of the respiratory epithelium, and is divided into two broad categories. Small cell lung cancers (SCLC) are a highly malignant tumor type derived from cells exhibiting neuroendocrine characteristics and account for approximately 15% of lung cancer cases.Citation3,Citation4 The remaining 85% of cases are non-small cell lung cancer (NSCLC), which is further divided into three major subtypes: squamous cell carcinoma, large cell carcinoma and adenocarcinoma. Although squamous cell carcinoma was previously the most frequently encountered histologic subtype across all ages, races and genders, in the last few decades it has been replaced by adenocarcinoma as the most prevalent type of NSCLC.Citation5,Citation6
The majority of lung cancers are directly attributable to tobacco smoking, which is associated with lung tumors of all major histological forms. However, this association is stronger for squamous cell carcinomas and small cell cancers than for adenocarcinoma.Citation4 Intriguingly a significant minority of lung cancer cases, the majority of which are adenocarcinomas, occurs in “never smokers”, often defined as those who have smoked fewer than 100 cigarettes in their lifetime. The percentage of lung cancers occurring in non-smokers appears to be increasing, with previous reports suggesting that 10% of malignancies were unconnected with smoking but more recent data giving a figure as high as 25%; indeed if lung cancer in never-smokers is considered in isolation it represents the seventh-most common cancer worldwide.Citation4,Citation7 The rise in incidence among non-smokers is not apparently explicable in terms of environmental tobacco smoke alone: although home or workplace exposure to tobacco smoke has been shown to lead to a modestly elevated risk of lung cancer, environmental smoke appears to be a relatively weak carcinogen in isolation.Citation8–Citation10 Indeed, given the differences in mutation signatures and responses to therapy between lung cancers in never-smokers versus smokers, it has been suggested that the two diseases are distinct entities.Citation11
The prognosis for lung cancer patients is poor, at least in part because NSCLC patients who make up the majority of cases are frequently diagnosed at an advanced stage of the disease.Citation2 This is reflected in the low five-year survival rates for lung cancer, at 15% in the United States, 10% across Europe and 9% in developing countries.Citation2 Whilst SCLC can be highly responsive to chemotherapy, offering the possibility of improved long-term survival when combined with radiotherapy, NSCLC remains considerably more refractory to treatment.Citation12 The aggressive and invasive nature of NSCLC leads to the establishment of distal metastases in organs including the bones, contralateral lung, liver and brain within months of diagnosis, and death in patients usually occurs due to this rapid metastatic spread rather than the primary lung tumors themselves.Citation13,Citation14 Patients diagnosed with early-stage NSCLC that has not yet metastasized to the lymph nodes or distal organs have a five-year survival of approximately 50%.Citation15 However, very few cases are diagnosed at this stage, the vast majority (around 85%) of patients presenting with advanced disease. The need for new and effective chemotherapeutic strategies to target advanced NSCLC is therefore a pressing one, and in the remainder of this review we will discuss the molecular origins of the disease with emphasis on its most common form, adenocarcinoma. Subsequently we will consider several recent publications which we believe offer exciting and potentially important insights into new opportunities for treatment.
Common Genetic Lesions in NSCLC
A variety of genetic abnormalities are encountered in lung cancer, with differing incidences in distinct histological subtypes of the disease; these were recently reviewed elsewhere.Citation3 Here we concentrate on two of the most common and widely studied genetic lesions in NSCLC, mutations of the epidermal growth factor receptor (EGFR) and KRAS genes.
EGFR.
EGFR, a member of the receptor tyrosine kinase (RTK) family, is a cell-surface receptor protein that responds to signals conveyed by extracellular growth factors. Following binding of these growth factor ligands, EGFR homo- or heterodimerizes with other RTKs and triggers the activation of a number of downstream signaling pathways with pleiotropic effects.Citation16 EGFR signaling to the phosphatidylinosityl 3-kinase-Akt (PI3KAkt) pathway leads to an inhibition of apoptosis and cell death. EGFR also mediates activation of the Ras-Raf-Mek-Erk signaling cascade, resulting in enhanced cellular proliferation, and stimulates STAT signaling, which in turn influences many other cellular processes that initiate increases in angiogenesis, invasion and metastasis.Citation4
Overexpression of the EGFR gene has been reported in 62% of NSCLC patients, and is associated with a poor prognosis.Citation17–Citation19 In some cases the canonical EGFR ligands epidermal growth factor (EGF) and transforming growth factorα (TGFα) may also be overexpressed, leading to EGFR hyperactivation through a signaling loop in which the ligands released by the cell further stimulate receptors on its own surface.Citation20,Citation21 Small molecule tyrosine kinase inhibitors (TKIs) have been developed to target EGFR, with gefitinib (Iressa, AstraZeneca) and erlotinib (Tarceva, OSI Pharmaceuticals, Genentech) approved by the US Food and Drug Administration for use in advanced cases of NSCLC refractory to conventional chemotherapeutic drugs.Citation22–Citation25 Early clinical trials gave modestly encouraging results, with partial responses to the TKI drugs observed in around 10% of NSCLC patients.Citation24,Citation26–Citation28 A small number of patients responded dramatically to TKI therapy, leading to the identification of mutations in the EGFR tyrosine kinase domain which are associated with drug sensitivity.Citation29,Citation30 The incidence of the different types of EGFR mutations in NSCLC is summarized in .
Kinase domain mutations are strongly associated with adenocarcinoma histology, patients who have never smoked, female gender and East Asian racial origin. In addition to TKI sensitivity, they are also associated with an improved response to conventional chemotherapeutic regimens.Citation16,Citation31–Citation33 Unfortunately, however, the bulk of patients who respond initially to erlotinib and gefitinib subsequently relapse.Citation34–Citation36 This resistance appears to occur through a range of different mechanisms. A T790M mutation was identified in exon 20 of the EGFR gene: this alters the binding kinetics of the “standard” TKI drugs, which bind reversibly to EGFR. However, a new generation of irreversible EGFR inhibitors, which have been shown to suppress T790M mutant tumor cells in vitro, may have potential as treatments for T790M mutant tumors.Citation34,Citation37,Citation38 Other proposed mechanisms of acquired TKI resistance include amplification of the MET proto-oncogene, activation of RTK family members other than EGFR, mutations in the KRAS gene, and epithelial-to-mesenchymal transition (EMT), although the underlying biological processes linking these events to drug resistance have not all been fully elucidated.Citation3,Citation35,Citation39–Citation43 Although success to date in targeting NSCLC with disregulated EGFR signaling has been limited, this is an area of intense research activity; two recent publications (among others) reported that combination therapy using both MET and EGFR inhibitors led to a pronounced reduction in carcinoma growth. The use of the anti-EGFR antibody cetuximab together with a second-generation EGFR TKI was also proposed as an effective strategy for overcoming T790M-mediated drug resistance.Citation44,Citation45 Efforts to develop new and effective drugs targeting EGFR-induced NSCLC will be greatly assisted by the development of transgenic mice harboring a range of different Egfr mutations that recapitulate different aspects of the disease. Mice with inducible tissue-specific expression of L858R or exon 19 deletion mutants of EGFR develop lung tumors that are susceptible to first-generation TKI therapy, whilst mice expressing the T790M mutant EGFR, either alone or in combination with an L858R mutation, develop drug-resistant tumors.Citation46,Citation47
KRAS.
Mutations of the RAS genes in lung cancer have a very different pattern of incidence from EGFR mutations (); they are entirely limited to NSCLC (predominantly adenocarcinoma), are much more common in neoplasms arising in smokers and less common in patients of East Asian origin.Citation3,Citation4 The majority of studies to date have not identified any gender bias in the incidence of KRAS mutations independent of tobacco smoking.Citation48 Although estimates vary somewhat, activating mutations in the KRAS gene account for 90% of mutations in the Ras pathway in lung cancer, and are reported to occur in 10–50% of human lung adenocarcinomas.Citation3,Citation35,Citation49–Citation53 In 97% of cases these mutations affect codons 12 or 13, and lead to a loss of the intrinsic Kras GTPase activity which is required to return the Ras protein to its inactive, GDP-bound state.Citation4,Citation51,Citation52,Citation54,Citation55 In consequence the negative feedback control of Ras activity is lost and the Ras-Raf-Mek-Erk kinase pathway constitutively activated, leading to uncontrolled cellular proliferation.
KRAS mutations are correlated with poor patient prognosis.Citation3 They are essentially mutually exclusive with EGFR mutations and are associated with resistance to both TKI therapy and conventional chemotherapy.Citation35,Citation51,Citation56 In contrast to EGFR mutant lung cancers, for which modestly effective chemotherapeutic drugs are available, with the prospect of more potent “second generation” therapies on the horizon, there is a lack of routinely effective targeted therapies against KRAS mutant lung adenocarcinoma. Given that this subtype makes up a significant proportion of all lung cancer cases, a robust research effort is currently directed towards understanding KRAS-induced lung tumors in more detail, with many studies using genetic mouse models that closely resemble the human disease. The “first generation” model involved expression of oncogenic KrasG12D as a “latent allele,” which was expressed spontaneously following recombination of the endogenous allele in somatic cells.Citation57,Citation58 Although a range of tumors and hyperplasias developed in these animals, the lung was the most sensitive organ to activated Kras expression, with the rapid onset of multiple adenocarcinomas and death from lung tumor burden.
Multiple “second generation” models have since been developed, in which the expression of activated Kras is spatially and temporally controlled using conditional alleles.Citation58 A widely used model incorporates an activated mutant Kras allele in the endogenous locus, the expression of which is under the control of a Lox-STOP-Lox (LSL) cassette; administration of Cre recombinase to the lungs of the mice through intranasal infection with an adenovirus results in loss of the STOP codon and activated Kras expression.Citation59 This model allows precise control of the timing of tumor initiation, as well as tumor multiplicity by varying the dose of adenovirus used. Further, it was shown to recapitulate closely the progression of human lung adenocarcinoma, from initial hyperplasia to adenomatous lesions and subsequently frank adenocarcinomas. Genetic models expressing activated Kras alone have one main deficiency, in that they typically do not exhibit invasion, stromal desmoplasia or metastasis; in contrast, over 50% of human lung cancers are diagnosed at an advanced stage with distal metastases.Citation58 In order to produce models that more closely model human adenocarcinoma and therefore provide an improved insight into potential therapies, a variety of improved systems has been developed by combining Kras activation with mutations or deletions of the tumor suppressor genes Trp53 and Lkb1.Citation15,Citation60,Citation61 These mice exhibit extremely aggressive and invasive disease, with a large stromal component and varying patterns of distant metastasis, representing an extremely useful tool for studying the advanced stages of human lung adenocarcinoma.
Recent Therapeutic Insights
Reliable and effective treatments for NSCLC are an urgent and currently unmet need. In the remainder of this review we will discuss a selection of very recent papers that offer potentially exciting therapeutic insights, with particular emphasis on KRAS mutant adenocarcinoma.
Synthetic lethal interactions.
A “synthetic lethal” interaction exists between two genes if mutations in either alone allow viability, but simultaneous mutation in both leads to death.Citation62 These interactions are potentially very valuable in cancer therapy, since targeting a gene that is synthetic lethal to a cancer-causing mutation should kill only cancer cells and not normal tissue, therefore reducing drug toxicity and side-effects. Recent work from the laboratory of Mariano Barbacid has suggested that one such interaction may exist between Kras and Cdk4, one of the cyclin-dependent kinase (Cdk) genes involved in cell cycle regulation.Citation63
Together the Cdk2, Cdk4 and Cdk6 genes are known as “interphase” Cdks, since they contribute to a number of cell cycle processes that occur during interphase.Citation64 Although these interphase Cdks appear to be dispensable for normal homeostasis in most mouse cells, they are required to drive proliferation of certain cell types at specific developmental stages.Citation64,Citation65 The authors investigated the possibility that these three Cdks might be selectively required by tumor cells depending on their cellular origin and pathogenic profile. Initial experiments in MEFs expressing a KrasG12V allele showed that loss of any of the three interphase Cdks compromised the proliferative advantage conferred by Kras activation. Knockdown of these genes using shRNA also led to a proliferation defect in human NSCLC cell lines harboring KRAS mutations. Following these in vitro studies the authors used mice carrying a conditional KrasG12V allele, and induced lung adenocarcinoma in the presence or absence of Cdk2, Cdk4 or Cdk6. Loss of Cdk6 had little effect on tumor progression in these animals. Although loss of Cdk2 led to an increase in survival and reduced tumor burden, the tumor stage profile remained essentially unchanged, suggesting that Cdk2 may inhibit the initiation of KRAS-dependent adenocarcinoma but not its progression. Strikingly, Cdk4 null mice showed a six- to eight-fold reduction in tumor burden, together with a dramatic retardation of tumor progression when compared to control animals. Puyol et al. demonstrated that this effect was due to the immediate onset of cellular senescence when KrasG12V was expressed specifically in Cdk4-/- lung tissue; this response was not observed in KrasG12V-expressing stomach, colon, pancreas or thymus. Importantly, experiments using mice with conditional alleles for both KrasG12V and Cdk4 demonstrated that acute deletion of Cdk4 inhibited the progression of established NSCLC, indicating that Cdk4 is essential for the progression of Kras-driven adenocarcinoma. Further, mice treated with PD0332991, an inhibitor of Cdk4 and Cdk6 that shows no significant activity against other Cdks, showed a retardation in disease progression compared to control-treated animals.Citation63,Citation66 The relationship that this work has established between Kras and Cdk4 is summarized in . These results raise the exciting possibility that a specific Cdk4 inhibitor could be a selective and effective drug to use against advanced NSCLC.
Personalized or tailored therapies.
Recent advances in genomic and proteomic sequencing technologies raise the possibility that molecular profiling may soon be an effective way to interrogate tumor genotypes and phenotypes, directing treatments that may be able to improve clinical outcomes. As a prelude to this approach, researchers at Genentech have recently obtained the complete sequence of a KRAS-mutant primary lung adenocarcinoma and adjacent normal tissue from a male smoker.Citation67 This approach allows an unbiased assessment of the mutation spectrum in a tumor, rather than relying on a limited set of previously-identified, commonly mutated genes. This work identified a wide range of somatic mutations, with a relatively higher rate of amino acid-changing mutations in kinase genes. Mutations or amplifications were detected in at least eight genes in the EGFR-Ras-Raf-Mek-Erk cascade, with multiple mutations also detected in other cancer-related signaling pathways. The authors conclude that tumors may therefore contain many different partially redundant mutations rather than being “addicted” to a single oncogene for survival. As sequencing technologies improve in the future, similar analyses in patient samples could prove instrumental in informing and customizing therapeutic strategies to give the maximum possible benefit.
Researchers at the Dana-Faber Cancer Institute in Boston have used a combined proteomic and genomic profiling approach to identify gene and phosphoprotein “signatures” associated with invasive and metastatic lung tumors.Citation68 This work utilized a transgenic model of Kras-induced adenocarcinoma which becomes invasive when combined with Lkb1 deletion, metastasizing to the lymph nodes and distal organs. Gene signatures for metastasis were identified by comparing mRNA expression profiles in primary tumors with those in both distal and lymph-node metastases. This signature was validated as a prognostic indicator in human NSCLC by comparison with similar data obtained from human samples. A proteomic analysis of changes in protein phosphorylation was also carried out to identify phosphorylated proteins that were enriched in the absence of Lkb1, leading to the identification of Src-family kinase activation as a key signaling pathway in Lkb1-deficient primary and metastatic lesions. Although previous work had shown that combined inhibition of PI3K-mTOR and Mek signaling resulted in an 80% reduction of tumor volume in mice with Kras-driven adenocarcinoma, the Lkb1-deficient tumors did not respond well to this therapy.Citation68,Citation69 However when a Src inhibitor was added to the dosing regimen, significant tumor regression was observed; further, although treatment with a Src inhibitor alone conferred no survival benefit it did block metastasis. These results provide an important proof of the principle that protein and gene profiling approaches have potential as a means to tailor cancer therapy.
Further evidence of a role for genetic analysis of tumors in informing cancer therapy comes from Carla Kim's research group at Harvard. This work has shown that the markers used to identify cancer stem cells vary depending on the primary tumor genotype.Citation70 In the cancer stem cell/tumor propagating cell (TPC) model, only a certain number of stem-like tumor cells possess the property of self-renewal and the capacity to propagate the tumor phenotype in vivo, leading to tumor initiation and distant metastases.Citation71 Populations of these TPCs have been identified in a range of cancers: in some cases virtually every cell in the tumor has the ability to propagate, in others the ratio of TPCs is much lower.Citation72,Citation73 In this work the researchers used three different transgenic murine NSCLC models, removed primary tumors by microdissection and after dissociating the cells transplanted them into the lungs of immunodeficient recipient mice in order to assay the self-renewing potential of the original tumors. Limiting dilution analysis revealed that around one cell in 10,000 gave rise to a new tumor, and that this number was approximately the same in two different models. Next the expression of Sca1, a marker of normal lung stem/progenitor cells, was analyzed. This resulted in a significant enrichment of TPCs in the Sca1+ population compared to the Sca1- cells in tumors with KrasG12D expression and p53 deletion. However, isolation of Sca1+ and Sca1− cells from tumors carrying the Kras mutation alone did not enrich TPCs in either population, indicating that in these tumors Sca1 was not a marker for “stemness.” Further, in a third model of EGFR-induced NSCLC, whilst around 1% of the cells were Sca1+, only the Sca1− population was capable of propagating EGFR tumors, indicating that TPCs in Kras-driven tumors are distinct from those in EGFR-induced disease. Since all cells capable of regenerating a tumor need to be eliminated or disabled for successful anti-cancer therapy, this work raises the possibility that the identification of TPCs in different types of NSCLC may in future enable specific targeting of these cells in different tumor types and patients.
Hypoxia.
Tissue hypoxia is generally defined as a partial pressure of oxygen equivalent to 15 mmHg or less. It is a common feature in regions of solid tumors, where it arises due to the disorganized vasculature, defective tumor vessel autoregulation and transient interruptions in blood supply that occur in these lesions.Citation74,Citation75 Transcriptional responses to hypoxia are mediated through a conserved signaling pathway involving the hypoxia inducible transcription factors (HIFs). These are αβ-heterodimers consisting of an oxygen-labile α-subunit which is degraded under normoxic conditions and an oxygen-insensitive β-subunit.Citation75–Citation78 This system is known to regulate approximately two-hundred genes, many of which are involved in processes relevant to cancer including energy metabolism, angiogenesis, cell growth, migration, invasion and metastasis.Citation74,Citation75 The two best-characterized HIFα proteins, HIF1α and HIF2α, have partially overlapping but distinct patterns of gene regulation. For reviews of HIF signaling, see references Citation74, Citation76 and Citation77.
Although the existence of tumor hypoxia in NSCLC may seem counterintuitive in the context of extensive oxygenation of lung tissue, overexpression of the HIF1α protein has been reported in 33–62% of NSCLCs, together with overexpression of HIF2α which is associated with a poor prognosis.Citation79,Citation80 Further, HIF2α is expressed at a relatively high level in type II pneumocytes, proposed as a cell of origin for NSCLC.Citation59,Citation81,Citation82 These data, combined with the observation that a monoclonal antibody targeting vascular endothelial growth factor (VEGF), a canonical HIF target gene, has shown some clinical benefits in NSCLC patients suggested a role for HIF signaling in lung cancer which has recently been investigated by two separate research teams.Citation83,Citation84 The first study, led by William Kim's laboratory at the University of North Carolina, investigated the effect of overexpressing a constitutively active HIF2α mutant concurrently with tumor activation in a conditional KrasG12D adenocarcinoma model.Citation85 HIF2α-expressing mice developed larger tumors and showed shortened survival, with distinct gene expression changes consistent with upregulation of canonical HIF targets. The HIF2α-expressing tumors were also more poorly differentiated and invasive when compared to their control counterparts. Based on analysis of an established set of marker genes, they showed a greater tendency towards EMT, a program of cell development involving loss of adhesion, E-cadherin repression and an increase in cell mobility. Further, a HIF2α “gene signature” associated with EMT was derived from the mouse tumors and validated using human clinical specimens. This demonstration that HIF2α could promote tumor growth in an autochthonous tumor model supports previous data implicating HIF2α in renal carcinoma and suggests a causal link between HIF2α expression and prognosis in NSCLC.
However, a second study conducted in our laboratory suggests that the role of HIF2α in NSCLC may not be as simple as it initially appeared. Although using the same Kras-induced adenocarcinoma model, our approach differed in that we employed conditional Hif1α and Hif2α alleles to delete these proteins concurrently with tumor initiation, rather than overexpressing them.Citation86 Surprisingly, in our hands deletion of HIF1α had little effect on the progress of NSCLC, but deletion of HIF2α led to an increase in tumor burden, proliferation and progression. Global gene expression profiling was carried out on tumors that were wild-type or null for HIF2α and revealed a small set of genes whose expression was reduced significantly in the absence of HIF2α. One of these genes was Scgb3a1, an intriguing target since its expression is primarily epithelial, it is silenced in a variety of human cancers and its downregulation is a significant predictor of poor clinical outcome in early-stage NSCLC.Citation87–Citation90 Further analysis revealed that Scgb3a1 was a novel target gene for HIF2α. Consistent with a previous report that forced expression of Scgb3a1 inhibited Akt activity in human breast cancer cells, HIF2α deletion led to increased Akt signaling, an effect that could be reversed in vitro by restoring either HIF2α or Scgb3a1 protein expression.Citation86,Citation91 Finally, we demonstrated that HIF2α and SCGB3A1 mRNA levels were correlated in human NSCLC.
The development and discovery of small-molecule inhibitors of the HIF pathway as potential chemotherapeutic agents is the subject of much current research.Citation92–Citation95 Taking our results together with those of Kim et al. it would appear that the inhibition of overexpressed HIF2α may be of clinical benefit in NSCLC (and perhaps other cancers), but that inhibition below a certain threshold could prove deleterious to the patient by inhibiting the expression of tumor suppressor genes (). As current HIF-inhibitory drugs do not appear to be selective for one isoform of HIF over another, work is continuing to investigate the potential clinical benefit to patients of pan-HIF inhibition as a chemotherapeutic strategy in lung cancer.
Conclusions
Lung cancer is likely to remain the foremost cause of cancer deaths for some time to come, as its treatment is an important clinical need that remains largely unmet. Although medicine is still a considerable distance from being able to treat all forms of the disease, and in particular the more lethal forms of NSCLC, we believe that recent research offers some cause for cautious optimism. Second-generation chemotherapeutic regimens which may have enhanced success in tackling EGFR-mutant cancers are in development. We have described a range of new insights, including the identification of synthetic lethal interactions, the use of genomics and proteomics to characterize tumors or TPCs and therefore direct treatment and the therapeutic possibilities the inhibition of the HIF pathway may offer in due course. Much work remains to be done, since many of these technologies and fields are still at a relatively early stage; however we believe that they offer exciting possibilities for the future in tackling this devastating disease.
Abbreviations
| SCLC | = | small cell lung cancer |
| NSCLC | = | non-small cell lung cancer |
| EGFR | = | epidermal growth factor receptor |
| RTK | = | receptor tyrosine kinase |
| TKI | = | tyrosine kinase inhibitor |
| Cdk | = | cyclin-dependent kinase |
| TPC | = | tumor propagating cell |
| HIF | = | hypoxia-inducible factor |
| EMT | = | epithelial-mesenchymal transition |
Figures and Tables
Figure 1 The interaction between Kras and Cdk4 genes in a mouse model of NSCLC. Although lung cells with Kras mutation or Cdk4 deletion alone are viable, the combination of both genetic alterations leads to immediate cellular senescence, potentially paving the way for a specific Cdk4 inhibitor to be used therapeutically.
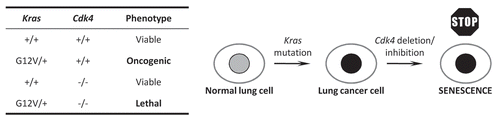
Figure 2 A model for the role of HIF2α in NSCLC. Both overexpression and deletion of HIF2α may promote tumor growth, with these effects occurring through the induction of protumorigenic target genes or the downregulation of tumor suppressor target genes respectively. Adapted from Mazumdar et al.Citation86
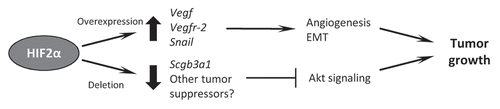
Table 1 Comparison of the incidence and characteristics of EGFR and KRAS mutations in human NSCLC
Acknowledgements
We thank Brian Keith for helpful comments on the manuscript. Work in the authors' laboratory is funded by the Howard Hughes Medical Institute (M.C.S.) and National Institutes of Health grant HL66130 (to B.K. and M.C.S.).
References
- Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of eighteen major cancers in 1985. Int J Cancer 1993; 54:594 - 606
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics 2002. CA Cancer J Clin 2005; 55:74 - 108
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008; 359:1367 - 1380
- Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers—a different disease. Nat Rev Cancer 2007; 7:778 - 790
- Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J 2001; 18:1059 - 1068
- Travis WD, Travis LB, Devesa SS. Lung cancer. Cancer 1995; 75:191 - 202
- Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer 2003; 3:733 - 744
- Tobacco smoke and involuntary smoking. IARC Monogr Eval Carcinog Risks Hum 2004; 83:1 - 1438
- Stayner L, Bena J, Sasco AJ, Smith R, Steenland K, Kreuzer M, et al. Lung cancer risk and workplace exposure to environmental tobacco smoke. Am J Public Health 2007; 97:545 - 551
- Vineis P, Alavanja M, Buffler P, Fontham E, Franceschi S, Gao YT, et al. Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst 2004; 96:99 - 106
- Toh CK, Gao F, Lim WT, Leong SS, Fong KW, Yap SP, et al. Never-smokers with lung cancer: epidemiologic evidence of a distinct disease entity. J Clin Oncol 2006; 24:2245 - 2251
- Hoffman PC, Mauer AM, Vokes EE. Lung cancer. Lancet 2000; 355:479 - 485
- Feld R, Rubinstein LV, Weisenberger TH. Sites of recurrence in resected stage I non-small-cell lung cancer: a guide for future studies. J Clin Oncol 1984; 2:1352 - 1358
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009; 9:274 - 284
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 2005; 65:10280 - 10288
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7:169 - 181
- Hirsch FR, Varella-Garcia M, Bunn PA Jr, Di Maria MV, Veve R, Bremmes RM, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 2003; 21:3798 - 3807
- Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001; 37:9 - 15
- Ohsaki Y, Tanno S, Fujita Y, Toyoshima E, Fujiuchi S, Nishigaki Y, et al. Epidermal growth factor receptor expression correlates with poor prognosis in non-small cell lung cancer patients with p53 overexpression. Oncol Rep 2000; 7:603 - 607
- Putnam EA, Yen N, Gallick GE, Steck PA, Fang K, Akpakip B, et al. Autocrine growth stimulation by transforming growth factoralpha in human non-small cell lung cancer. Surg Oncol 1992; 1:49 - 60
- Rusch V, Baselga J, Cordon-Cardo C, Orazem J, Zaman M, Hoda S, et al. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res 1993; 53:2379 - 2385
- Blackhall F, Ranson M, Thatcher N. Where next for gefitinib in patients with lung cancer?. Lancet Oncol 2006; 7:499 - 507
- Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 2005; 23:5892 - 5899
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005; 353:123 - 132
- Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366:1527 - 1537
- Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol 2003; 21:2237 - 2246
- Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290:2149 - 2158
- Perez-Soler R, Chachoua A, Hammond LA, Rowinsky EK, Huberman M, Karp D, et al. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. J Clin Oncol 2004; 22:3238 - 3247
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 2139
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 1500
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008; 358:1160 - 1174
- Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet 2008; 372:1809 - 1818
- Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer 2006; 118:257 - 262
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:73
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2:17
- Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol 2007; 25:587 - 595
- Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 2006; 66:7854 - 7858
- Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 2005; 102:7665 - 7670
- Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 2007; 104:20932 - 20937
- Cappuzzo F, Janne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol 2009; 20:298 - 304
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316:1039 - 1043
- Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res 2007; 13:2795 - 2803
- Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 2005; 11:8686 - 8698
- Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 2009; 119:3000 - 3010
- Zhang YW, Staal B, Essenburg C, Su Y, Kang L, West RA, et al. MET kinase inhibitor SGX523 synergizes with EGFR inhibitor erlotinib in an HGF-dependent fashion to suppress carcinoma growth. Cancer Res 2010; 70:6880 - 6890
- Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to downregulation of the receptors. Genes Dev 2006; 20:1496 - 1510
- Regales L, Balak MN, Gong Y, Politi K, Sawai A, Le C, et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One 2007; 2:810
- Gazdar AF, Thun MJ. Lung cancer, smoke exposure and sex. J Clin Oncol 2007; 25:469 - 471
- Kobayashi T, Tsuda H, Noguchi M, Hirohashi S, Shimosato Y, Goya T, et al. Association of point mutation in c-Ki-ras oncogene in lung adenocarcinoma with particular reference to cytologic subtypes. Cancer 1990; 66:289 - 294
- Mills NE, Fishman CL, Rom WN, Dubin N, Jacobson DR. Increased prevalence of K-ras oncogene mutations in lung adenocarcinoma. Cancer Res 1995; 55:1444 - 1447
- Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res 2008; 14:5731 - 5734
- Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc 2009; 6:201 - 205
- Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, et al. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res 1988; 48:5738 - 5741
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res 1989; 49:4682 - 4689
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3:11 - 22
- Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005; 23:5900 - 5909
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001; 410:1111 - 1116
- Kim CF, Jackson EL, Kirsch DG, Grimm J, Shaw AT, Lane K, et al. Mouse models of human non-small-cell lung cancer: raising the bar. Cold Spring Harb Symp Quant Biol 2005; 70:241 - 250
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001; 15:3243 - 3248
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007; 448:807 - 810
- Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene 2007; 26:6896 - 6904
- Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5:689 - 698
- Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010; 18:63 - 73
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153 - 166
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007; 448:811 - 815
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3:1427 - 1438
- Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010; 465:473 - 477
- Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell 2010; 17:547 - 559
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008; 14:1351 - 1356
- Curtis SJ, Sinkevicius KW, Li D, Lau AN, Roach RR, Zamponi R, et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 2010; 7:127 - 133
- Sullivan JP, Minna JD. Tumor oncogenotypes and lung cancer stem cell identity. Cell Stem Cell 2010; 7:2 - 4
- Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 2009; 138:822 - 829
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8:755 - 768
- Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer 2008; 8:967 - 975
- Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008; 30:393 - 402
- Lisy K, Peet DJ. Turn me on: regulating HIF transcriptional activity. Cell Death Differ 2008; 15:642 - 649
- Webb JD, Coleman ML, Pugh CW. Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell Mol Life Sci 2009; 66:3539 - 3554
- Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007; 2007:8
- Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Talks K, Pezzella F, et al. Relation of hypoxia inducible factor1alpha and 2alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br J Cancer 2001; 85:881 - 890
- Kim SJ, Rabbani ZN, Dewhirst MW, Vujaskovic Z, Vollmer RT, Schreiber EG, et al. Expression of HIF-1alpha, CA IX, VEGF and MMP-9 in surgically resected non-small cell lung cancer. Lung Cancer 2005; 49:325 - 335
- Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 2002; 8:702 - 710
- Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA 1997; 94:4273 - 4278
- Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006; 355:2542 - 2550
- Socinski MA, Novello S, Brahmer JR, Rosell R, Sanchez JM, Belani CP, et al. Multicenter, phase II trial of sunitinib in previously treated, advanced non-small-cell lung cancer. J Clin Oncol 2008; 26:650 - 656
- Kim WY, Perera S, Zhou B, Carretero J, Yeh JJ, Heathcote SA, et al. HIF2alpha cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest 2009; 119:2160 - 2170
- Mazumdar J, Hickey MM, Pant DK, Durham AC, Sweet-Cordero A, Vachani A, et al. HIF-2alpha deletion promotes Kras-driven lung tumor development. Proc Natl Acad Sci USA 2010; 107:14182 - 14187
- Krop I, Player A, Tablante A, Taylor-Parker M, Lahti-Domenici J, Fukuoka J, et al. Frequent HIN-1 promoter methylation and lack of expression in multiple human tumor types. Mol Cancer Res 2004; 2:489 - 494
- Marchetti A, Barassi F, Martella C, Chella A, Salvatore S, Castrataro A, et al. Downregulation of high in normal-1 (HIN-1) is a frequent event in stage I non-small cell lung cancer and correlates with poor clinical outcome. Clin Cancer Res 2004; 10:1338 - 1343
- Porter D, Lahti-Domenici J, Torres-Arzayus M, Chin L, Polyak K. Expression of high in normal-1 (HIN-1) and uteroglobin related protein-1 (UGRP-1) in adult and developing tissues. Mech Dev 2002; 114:201 - 204
- Shigematsu H, Suzuki M, Takahashi T, Miyajima K, Toyooka S, Shivapurkar N, et al. Aberrant methylation of HIN-1 (high in normal-1) is a frequent event in many human malignancies. Int J Cancer 2005; 113:600 - 604
- Krop I, Parker MT, Bloushtain-Qimron N, Porter D, Gelman R, Sasaki H, et al. HIN-1, an inhibitor of cell growth, invasion and AKT activation. Cancer Res 2005; 65:9659 - 9669
- Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA 2009; 106:2353 - 2358
- Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth and vascularization. Proc Natl Acad Sci USA 2009; 106:17910 - 17915
- Shin DH, Kim JH, Jung YJ, Kim KE, Jeong JM, Chun YS, et al. Preclinical evaluation of YC-1, a HIF inhibitor, for the prevention of tumor spreading. Cancer Lett 2007; 255:107 - 116
- Won MS, Im N, Park S, Boovanahalli SK, Jin Y, Jin X, et al. A novel benzimidazole analogue inhibits the hypoxia-inducible factor (HIF)-1 pathway. Biochem Biophys Res Commun 2009; 385:16 - 21