Abstract
The role of autophagy in tumorigenesis is controversial. Both autophagy inhibitors (chloroquine) and autophagy promoters (rapamycin) block tumorigenesis by unknown mechanism(s). This is called the “Autophagy Paradox”. We have recently reported a simple solution to this paradox. We demonstrated that epithelial cancer cells use oxidative stress to induce autophagy in the tumor microenvironment. As a consequence, the autophagic tumor stroma generates recycled nutrients that can then be used as chemical building blocks by anabolic epithelial cancer cells. This model results in a net energy transfer from the tumor stroma to epithelial cancer cells (an energy imbalance), thereby promoting tumor growth. This net energy transfer is both unilateral and vectorial, from the tumor stroma to the epithelial cancer cells, representing a true host-parasite relationship. We have termed this new paradigm “The Autophagic Tumor Stroma Model of Cancer Cell Metabolism” or “Battery-Operated Tumor Growth”. In this sense, autophagy in the tumor stroma serves as a “battery” to fuel tumor growth, progression, and metastasis, independently of angiogenesis. Using this model, the systemic induction of autophagy will prevent epithelial cancer cells from using recycled nutrients, while the systemic inhibiton of autophagy will prevent stromal cells from producing recycled nutrients—both effectively “starving” cancer cells. We discuss the idea that tumor cells could become resistant to the systemic induction of autophagy, by the up-regulation of natural endogenous autophagy inhibitors in cancer cells. Alternatively, tumor cells could also become resistant to the systemic induction of autophagy, by the genetic silencing/deletion of pro-autophagic molecules, such as Beclin1. If autophagy resistance develops in cancer cells, then the systemic inhibition of autophagy would provide a therapeutic solution to this type of drug resistance, as it would still target autophagy in the tumor stroma. As such, an anti-cancer therapy that combines the alternating use of both autophagy promoters and autophagy inhibitors would be expected to prevent the onset of drug resistance. We also discuss why anti-angiogenic therapy has been found to promote tumor recurrence, progression, and metastasis. More specifically, anti-angiogenic therapy would induce autophagy in the tumor stroma via the induction of stromal hypoxia, thereby converting a non-aggressive tumor type to a “lethal” aggressive tumor phenotype. Thus, uncoupling the metabolic parasitic relationship between cancer cells and an autophagic tumor stroma may hold great promise for anti-cancer therapy. Finally, we believe that autophagy in the tumor stroma is the local microscopic counterpart of systemic wasting (cancer-associated cachexia), which is associated with advanced and metastatic cancers. Cachexia in cancer patients is not due to decreased energy intake, but instead involves an increased basal metabolic rate and increased energy expenditures, resulting in a negative energy balance. Importantly, when tumors were surgically excised, this increased metabolic rate returned to normal levels. This view of cachexia, resulting in energy transfer to the tumor, is consistent with our hypothesis. So, cancer-associated cachexia may start locally as stromal autophagy, and then spread systemically. As such, stromal autophagy may be the requisite precursor of systemic cancer-associated cachexia.
Acknowledgements
M.P.L. and his laboratory were supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660) and the Susan G. Komen Breast Cancer Foundation. F.S. was supported by grants from the W.W. Smith Charitable Trust, the Breast Cancer Alliance (BCA) and a Research Scholar Grant from the American Cancer Society (ACS). A.K.W. was supported by a Young Investigator Award from Breast Cancer Alliance, Inc. and a Susan G. Komen Career Catalyst Grant. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant from the Pennsylvania Department of Health (to M.P.L.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Figures and Tables
Figure 1 Prognostic value of caveolin-1 (Cav-1) as a stromal biomarker for DCIS, breast cancer and prostate cancer. (A) Schematic diagram summarizing that a loss of stromal Cav-1 is a new biomarker for hypoxia, oxidative stress, autophagy and the “Reverse Warburg Effect.” Importantly, a loss of stromal Cav-1 effectively predicts poor clinical outcome in DCIS (ductal carcinoma in situ), breast cancer and prostate cancer patients. Given that most human tumors have a stromal component, a loss of stromal Cav-1 may have prognostic value in a wide variety of different types of human cancers. Thus, a loss of stromal Cav-1 could be used to identify high-risk cancer patients at diagnosis, facilitating treatment stratification and clinical management. LVI, lympho-vascular invasion. (B) Kaplan-Meier analysis of stromal Cav-1 predicts overall survival in triple negative (TN) breast cancer patients. Patients with high levels of stromal Cav-1 (score = 2) had a good clinical outcome, with 75.5% of the patients remaining alive during the follow-up period (nearly 12 years). In contrast, the median survival for patients with absent stromal Cav-1 staining (score = 0) was 25.7 months. The results of this analysis were highly statistically significant (p = 2.8 × 10−6). (B) was reproduced from reference Citation8, with permission from the publisher (Landes Bioscience).
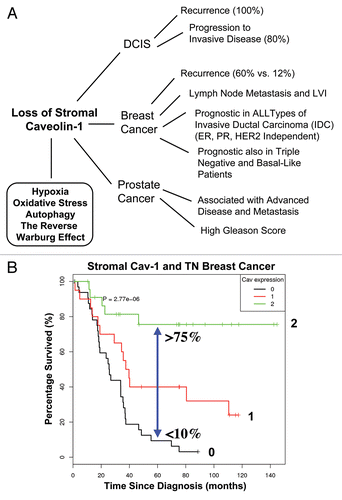
Figure 2 The autophagic tumor stroma model of cancer metabolism: Role of oxidative stress, recycled nutrients and random mutagenesis. (A) Oxidative Stress, Recycled Nutrients and Resistance to Apoptosis. We have shown that human breast cancer cells induce ROS (reactive oxygen species) production in adjacent cancer-associated fibroblasts (CAFs), leading to the onset of stromal oxidative stress. Oxidative stress in CAFs, in turn, drives autophagy via HIF1 induction and NFκB activation, resulting in the autophagic destruction of mitochondria (mitophagy) and caveolin-1 (Cav-1). Stromal autophagy and mitophagy provides recycled nutrients via catabolism and aerobic glycolsysis to feed the appetite of adjacent cancer cells. These recycled chemical building blocks (derived from autophagy and aerobic glycolysis) “fuel” oxidative mitochondrial metabolism in cancer cells and provide resistance against apoptosis. Mechanistically, fibroblasts induce the expression of TIGAR in adjacent cancer cells, which shuts down autophagy and apoptosis in these cancer cells. (B) Random mutagenesis and tumor-stroma co-evolution. Oxidative stress in cancer-associated fibroblasts (CAFs) also produces a local DNA damage response, with increased ROS production. ROS amplification in the tumor stroma then leads to an antioxidant defense in adjacent cancer cells. In addition, stromal ROS production also leads to DNA damage in adjacent cancer cells via a “Bystander Effect”. As a consequence, stromal ROS promotes aneuploidy and genomic instability in cancer cells, driving tumor-stroma co-evolution. CAFs, cancer-associated fibroblasts; ROS, reactive oxygen species.
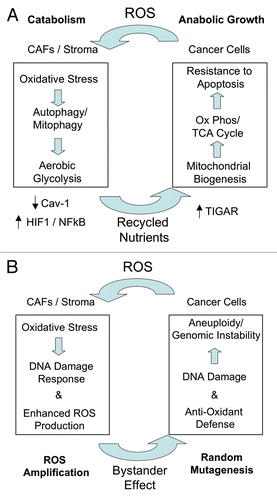
Figure 3 Understanding energy transfer in cancer metabolism: Stromal autophagy and cancer-associated cachexia. Diagram summarizing that autophagy in the tumor stroma is used by adjacent epithelial cells to fuel tumor growth via oxidative mitochondrial metabolism. This local phenomenon may spread systemically and explains the onset of cancer-associated cachexia, characterized by systemic wasting and a negative net energy balance. A+ signifies stromal autophagy, which results in fuel production. A− signifies an absence of autophagy in cancer cells. AR signifies genetic resitance to autophagy in cancer cells, e.g., when Beclin1 gene expression is silenced. Both A− and AR would be associated with fuel consumption by cancer cells. The direction of energy flow, from the autophagic tumor stroma to the anabolic cancer cells, is indicated by the direction of a blue arrow.
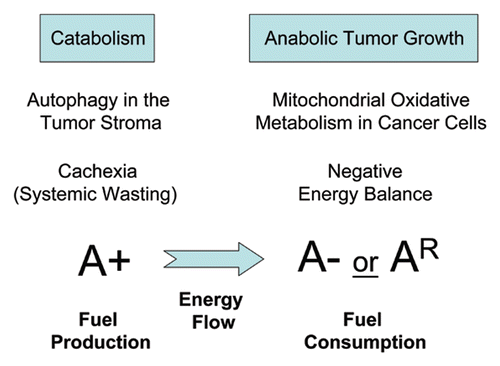
Figure 4 A simple solution to the autophagy paradox: “Battery Operated Tumor Growth”. The role of autophagy in tumorigenesis is controversial. Both autophagy inhibitors and autophagy promoters block tumorigenesis. This is called the “Autophagy Paradox”. We have reported a simple solution to this paradox. Mechanistically, we have shown that autophagy/catabolism in the tumor stroma drives the anabolic growth of adjacent cancer cells and tumor progression. In this sense, autophagy in the tumor stroma serves a “battery” to fuel tumor growth, progression and metastasis, independently of angiogenesis. Using this model, systemic induction of autophagy (with rapamycin) will prevent epithelial cancer cells from using recycled nutrients, while the systemic inhibiton of autophagy (with chloroquine) will prevent stromal cells from producing recycled nutrients—both effectively “starving” cancer cells. Tumor cells could become resistant to the systemic induction of autophagy by the upregulation of natural endogenous autophagy inhibitors in cancer cells (such as TIGAR). Alternatively, tumor cells could also become resistant to the systemic induction of autophagy by the genetic silencing of pro-autophagic molecules, such as Beclin1. If this occurs, then the systemic inhibition of autophagy would provide a therapeutic solution to this new type of drug resistance. CAFs, cancerassociated fibroblasts.
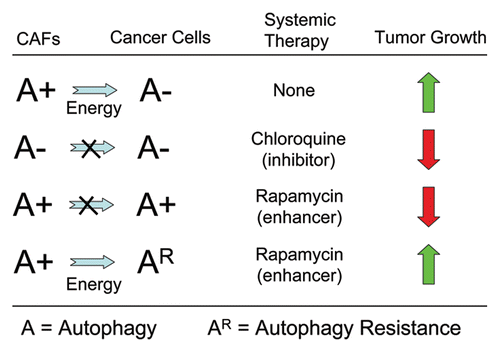
Figure 5 Understanding how anti-angiogenic therapy increases tumor progression, recurrence and metastasis via hypoxia in the tumor stroma. (A) Flow diagram summarizing how anti-angiogenic therapy drives a hypoxic/autophagic response in the tumor stromal micro-environment, which experimentally leads to tumor progression, recurrence and metastasis. This view is also supported by the lack of efficacy of anti-angiogenesis inhibitors in a variety of clinical trials in humans. (B) Converting a non-aggressive tumor to a “lethal” tumor via anti-angiogenic therapy. We have shown that autophagy in the tumor stroma fuels the anabolic growth of cancer cells and tumor progression. Thus, a non-aggressive tumor would lack stromal autophagy. This premise is supported by the use of stromal Cav-1 as a biomarker of autophagy. High stromal Cav-1 would be predictive of an absence of stromal autophagy and good clinical outcome. Conversely, low or absent stromal Cav-1 would be predictive of a high rate of stromal autophagy and poor clinical outcome. This is what we observed experimentally in our pre-clinical models and in our translational biomarker studies. Based on our model of “Battery-Operated Tumor Growth,” anti-angiogenic therapy would induce hypoxia and autophagy in the tumor stromal micro-enviroment, promoting net energy transfer and tumor progression and leading to a “lethal” tumor phenotype.
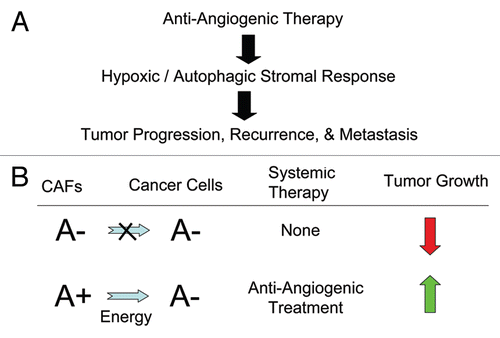
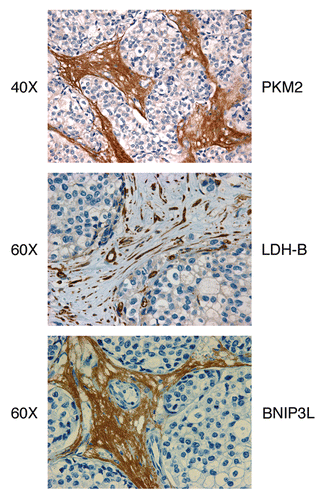
Figure 6 Aerobic glycolysis and mitophagy in the tumor stromal compartment. PKM2, LDH-B and BNIP3L are all highly expressed in the tumor stromal compartment of human breast cancers that lack the stromal expression of Cav-1. PKM2 and LDH-B are markers of aerobic glycolysis, while BNIP3L is a functional marker of the autophagic destruction of mitochondria (mitophagy). Images were reproduced from references Citation4, Citation26 and Citation27, with permission from the publisher (Landes Bioscience).