Abstract
Comment on: Ertel A, et al. Cell Cycle 2010; 9:4153–63.
The RB pathway is one of the best-studied and most frequently altered pathways leading to loss of proliferative control in cancer. It can be disrupted by a variety of means including, but not limited to, epigenetic silencing, allelic loss or mutation of RB1. In some cancers including breast cancer, amplification of the CCND1 gene or overexpression of cyclin D1 is commonly observed.Citation1 In others, the CDK4 gene is amplified or mutated within the p16INK4A-binding domain. Finally, many cancers lack p16INK4 expression due to deletion or promoter hypermethylation of CDKN2A. By increasing cyclin D1-CDK4 kinase activity, these genetic events all restrict the ability of underphosphorylated RB to inhibit cell cycle progression.
Genetic alterations at the RB1 locus, or lack of RB expression, occur in up to a third of breast cancers.Citation2 Breast cancer can be divided into a number of biologically distinct phenotypes, and RB loss is more common in some of these subtypes, i.e. basal breast cancers, which are typically estrogen receptor (ER)-negative, poorly-differentiated and aggressive and luminal B cancers, which are a poor-prognosis subgroup accounting for approximately a third of ER-positive breast cancers.Citation3 Interestingly, RB deletion in mouse mammary progenitor cells leads to the formation of tumors with basal or luminal B characteristics,Citation4 suggesting that RB loss may not simply be a marker of these subtypes.
Confounding issues in examining the association between RB loss and breast cancer therapeutic responsiveness and outcome include the poor correlation between RB1 LOH and low or absent RB protein expressionCitation5 as well as technical difficulties in measuring RB expression by immunohistochemistry. In addition, cyclin D1 overexpression or p16INK4A methylation, both of which are common in breast cancerCitation1,Citation6, are usually mutually exclusive with RB loss but are expected to have similar biological consequences. In a previous issue of Cell Cycle, Ertel et al.Citation7 used a “RB-loss signature” composed of genes that are upregulated by RB deletion or repressed by RB activation to identify breast cancers where the RB pathway is deregulated. In ER-positive cancers the RB-loss signature was correlated with cyclin D1 overexpression and low RB1 levels but not with p16INK4A levels, whereas it was correlated with p16INK4A/CDKN2A expression but not cyclin D1 or RB expression in ER-negative cancer. Although aberrant expression of these genes is to some degree a marker of RB pathway deregulation, the correlation is not strong, and none are useful biomarkers of pathway activity in all breast cancer subtypes.
Although in both ER-positive and ER-negative breast cancer, RB pathway deregulation was associated with increased proliferation, it was correlated with poorer outcome in ER-positive disease but with better outcome in ER-negative disease.Citation7 In ER-positive breast cancer this relationship was apparent both in patients treated with surgery alone and in patients receiving adjuvant antiestrogen (tamoxifen) therapy, indicating that ER-positive breast cancers with RB pathway deregulation are inherently more aggressive. They are also more likely to metastasise, possibly because of the decreased expression of cell-cell communication genes that Ertel et al. find to be associated with the RB-loss signature.Citation7 These observations are consistent with the widely-held view that more proliferative cancers have a poorer outcome, and the association between markers of increased proliferation and poor response to endocrine therapy.Citation8
In ER-negative cases, the association of RB pathway deregulation with better outcome appears to be due to improved response to some types of chemotherapy.Citation7,Citation9 This is likely to result, at least in part, from an impaired cellular DNA damage/genotoxic stress response in the absence of functional RB.Citation2 In ER-positive cancers, RB pathway deregulation may be a marker of increased sensitivity to small molecule inhibitors of CDK4.Citation7 These important findings provide impetus for further studies aimed at developing a clinically useful panel of biomarkers that can identify cancers with RB pathway deregulation and sensitivity to new therapies following independent validation in other patient cohorts. The importance of Ertel et al.'s work lies not only in that it points towards a potential role for measuring RB pathway aberrations in order to inform treatment decisions in breast cancer. It also illustrates the power of using gene signatures based on biological function to probe determinants of prognosis and response to therapy, as has been suggested in the context of endocrine therapy.Citation8
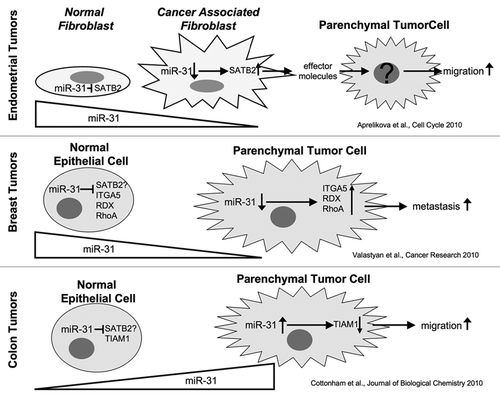
In epitheial tumors, a significant part of the tumor mass is comprised of non-parenchymal cells such as immune cells, cells of the microvasculature and fibroblasts, often referred to as tumor stroma or tumor microenvironment. The tumor stroma not only supplies scaffolding and nutrition to parenchymal tumor cells, but stromal and parenchymal cells also partake in an extensive crosstalk that can alter tumor biology. Fibroblasts are ubiquitous stromal cells that secrete extracellular matrix, such providing mechanical support to parenchymal cells. In tumors, cancer associated fibroblasts (CAFs) overproduce extracellular matrix thus contributing to the rigidity of tumors. Furthermore, CAFs secrete cytokines and growth factors into the tumor microenvironment thus promoting tumor growth and/or metastases.Citation1,Citation2
MicroRNAs (miRNAs) are small, non-coding, single-stranded RNAs that negatively regulate gene expression on a post-transcriptional level. Individual miRNAs can target multiple genes and such orchestrate complex biological phenotypes like cell migration or proliferation. On the other side, alterations of miRNA expression patterns might cause significant disturbances of cell and tissue homeostasis. Indeed, recently altered miRNA expression has been associated with cardiovascular diseases, diabetes and metabolic diseases, neurodenerative diseases and cancer.Citation3
In their present study, Aprelikova et al.Citation4 set out to test if posttranscriptional regulation of gene expression in endometrial CAFs as compared to endometrial normal fibroblasts (NFs) isolated from the same patient contributes to tumor progression. Having ascertained differential effects of CAFs and matching NFs on endometrial tumor cells in a xenograft model, Aprelikova et al.Citation4 determined which miRNAs and mRNAs were differentially expressed in CAFs and NFs and found miR-31 was the most downregulatated miRNA in CAFs, while one of its target genes, SATB2, was upregulated in CAFs. Release of miR-31 relayed suppression of SATB2 expression in fibroblasts increased migration of tumor cells towards fibroblasts in vitro while there was no effect on tumor cell proliferation. This indicates that expression levels of miR-31/SATB1 in stromal cells might influence the metastatic capacity of endometrial tumors.
Altered expression levels of miR-31 and SATB2 have been shown for several solid tumors. Interestingly, miR-31 expression is downregulated in carcinoma of the breast, prostate, ovary and stomach, but upregulated colorectal, liver and head-and-neck tumors.Citation5 Likewise, increased expression of SATB2 as shown to correlate with higher tumor grade in breast cancer and reduced expression of SATB2 to correlate with higher tumor stage and metastasis in colon cancer.Citation6,Citation7
These results together imply that metastasis is affected by stromal as well as parenchymal expression of miR-31 and its target gene SATB2. However, the downstream effects of altered mi-R31/SATB2 expression must be quite different for stromal cells, parenchymal breast cancer cells derived from breast cancer and parenchymal colon cancer cells, respectively. In parenchymal breast cancer cells the effect of miR-31 on metastases can be phenocopied by concurrent downregulation of the miR-31 target genes integrin α5, radixin and rhoA,Citation8 an in colon cancer cells miR-31 attentuates expression of TIAM1 which is involved in regulation of cell motility;Citation9 This implies that miR-31 affects the metastatic capacity of parenchymal cells by directly altering their biological properties. In contrast, Aprelikova et al.Citation4 demonstrated that altered miR-31/SATB2 levels in fibroblasts alter migration of tumor cells that were separated from the fibroblasts by the membrane of a transwell chamber, that is without direct contact between fibroblasts and tumor cells. The effect of altered fibroblast miR-31/SATB2 levels on tumor cells therefore has to be relayed by soluble factors that are released from the fibroblasts into the surrounding microenvironment and subsequently alter behavior of neighboring tumor cells. Thus the data of Aprelikova et al.Citation4 adds a new perspective to the tissue- and cell specific role of miR-31/SATB2 tumor-stromal interactions, tumor cell migration and metastasis.
Figures and Tables
Figure 1 Effects of miR-31 on tumor cell migration and metastasis are cell type and tissue dependent.
References
- Proia DA, et al. Cell Cycle 2005; 4:1022 - 1025
- Bhowmick NA, et al. Science 2004; 303:848 - 851
- Kanwar JR, et al. Front Biosci (Schol Ed) 2:1113 - 1126
- Aprelikova O, et al. Cell Cycle 2010; 9:4387 - 4398
- Valastyan S, et al. Cell Cycle 9:2124 - 219
- Patani N, et al. Cancer Cell Int 2009; 9:18
- Wang S, et al. J Pathol 2009; 219:114 - 122
- Valastyan S, et al. Cancer Res 70:5147 - 5154
- Cottonham CL, et al. J Biol Chem 2010; 285:35293 - 35302
The nucleolus is the most obvious and best-studied structure within the nucleus. It has central roles in cellular metabolism, aging and the cell cycle. However, the repetitive nature of the DNA skeleton (rDNA) that forms the nucleolar core makes it recalcitrant to detailed structural resolution. Recent methodological advances have provided tantalizing insights into the structure of this most enigmatic organelle.
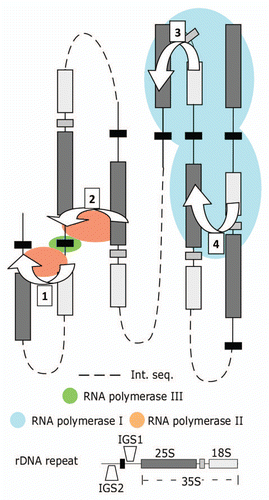
In yeast, the rDNA locus is made up of ∼200 repeats each consisting of the 35S rDNA (i.e., the 18S, 5.8S, 28S and intervening sequences) and 5S rDNA genes and two intergenic sequences (i.e., IGS1, IGS2). Inter- and intra-rDNA repeat interactions have been proposed as part of a structure that increases local concentrations of the cis- and trans-acting factors, DNA and protein, that are required for efficient rDNA transcription and replication.Citation1,Citation2 While theoretically attractive, these conformations have not been empirically confirmed.
In a previous issue, Mayan and AragónCitation3 presented suggestive observations relating to the structure of the yeast rDNA array. They identified an interaction that involves sequences either side of the 5S rDNA (IGS1-IGS2) and, while it is weak, seems to connect yeast 35S rDNA promoter and terminator regions. This interaction is similar to previously identified interactions between the 35S rDNA promoter and terminator regions in ratsCitation4 and mice.Citation5 Perhaps more importantly, there is also clear evidence for connections between IGS1 and the 25S rDNA and between regions located within the 18S and 25S rDNA genes. The IGS1-25SrDNA connection is consistent with an interaction we have previously documented in the yeast rDNA.Citation6 Together these results appear to substantiate earlier assertions that the terminal regions (promoters and terminators) of yeast rDNA repeats are connected.Citation1,Citation2 However, interpreting the results of proximity-based ligation assays of repetitive regions (e.g., the nucleolus) is particularly problematic because interactions that form within a repeat cannot be differentiated from those that form between different repeats (). Hence, it remains an open question as to whether these are intra-Citation1 or inter-repeatCitation2 interactions.
Mayan and Aragón make the intriguing observation that reducing the level of Reb1p (and to a lesser degree the Net1p) causes a reduction in the frequency of the IGS1-IGS2 interaction, and also of the interaction bridging the 18S and 25S genes.Citation3 Thus it appears that there is an inter-relationship between these two interactions. The link may be mediated by an interaction that occurs between the 25S rDNA and the enhancer/IGS1 sequence, which also requires enhancer and replication fork binding proteins (i.e., Fob1p and Sir2p).Citation6
Taken together with previous observations,Citation1,Citation2,Citation6 the results of Mayan and Aragón offer a suggestive glimpse of the spatial organization of the S. cerevisiae rDNA array (). Logically this organization facilitates reciprocal control of RNA polymerase I and cryptic RNA polymerase II transcription (e.g., TAR1) in the rDNA. Simply, these interactions can be invoked to establish mutually exclusive RNA polymerase I and II transcription factories between the 25S rDNA, promoter and terminator regions (). Other questions, including whether RNA polymerase III transcription factories are promoted by the formation of RNA polymerase I and/or II factories (Fig. 1.1 and 1.2) remain unanswered. This is particularly important because S. cerevisiae is one of the few organisms in which the 5S rDNA is found within the 35S rDNA repeats, therefore the formation of RNA polymerase III transcription factories may differ from those found in other eukaryotes. Moreover, there is no evidence as to whether the interactions require active RNA polymerase I transcription, despite tantalizing evidence that active RNA polymerase II is required (ref. Citation6, Mayan and Aragón). Finally, the dynamic nature of the nucleolus implicates systems that prevent and resolve nucleic acid knots in the maintenance of nucleolar structure. How these resolving systems relate to the structures that promote and stabilize spatial proximity within the rDNA remains to be determined. In short, it appears that our understanding of nucleolar structure will remain jumbled until the development and implementation of methods that enable the incremental untangling of individual rDNA repeats, and hence of the nucleolus itself.
Figures and Tables
Figure 1 Model for mutually exclusive formation of RNA polymerase I, II and III transcription factories within the yeast rDNA. Interactions bridging the IGS1-IGS2 (1) and the 25S rDNA-IGS1 (2) sequences form part of an RNA polymerase II transcription factory promoting transcription from cryptic RNA polymerase II promoters. These interactions isolate active 5S rDNA in yeast. The 25S - IGS1 interaction is mutually exclusive with the intra-35S (18-25S) rDNA loop (3) that positions IGS1 and IGS2 sequences for rapid re-initiation of RNA polymerase I and requires Rep1p binding and blockage at the replication fork binding site. For simplicity figures are depicted in two-dimensions, in reality the structures would form rosettes that merge the two halves (1, 2) of the polymerase II factories. The rosette structure would also merge the RNA polymerase I factories (3, 4). Proximity-based ligation methods are unable to differentiate interactions occurring within a repeat (3) from those occurring between repeats (4), yet they are structurally different. Int. seq.: intervening sequence that could include no, one or several rDNA repeats.
References
- Kempers-Veenstra AE, et al. EMBO J 1986; 5:2703 - 2710
- Johnson SP, et al. Mol Cell Biol 1989; 9:4986 - 4993
- Mayan M, et al. Cell Cycle 2010; 9:4328 - 4337
- Shiue CN, et al. Oncogene 2009; 28:1833 - 1842
- Nemeth A, et al. EMBO J 2008; 27:1255 - 1265
- O'Sullivan J, et al. Yeast 2009; 26:125 - 138
There is much to be said about keeping things simple even in this fast paced omics age. Just when we thought that for mapping global genetic networks, robotic systematic genetic analysis (SGA)Citation1 had replaced classical bench top random mutagenesis screens,Citation2,Citation3 Ma et al.Citation4 showed otherwise in a previous issue of Cell Cycle. In an effort to identify new players in the initiation of DNA replication, these authors used a phenotypic color-sectoring assay to screen EMS mutagenized cells for initiation of DNA replication (idr) mutants. A comparison of the results from this study and from a parallel SGA screenCitation5 yields an overlapping set of putative idr mutants but contrasting conclusions. Both studies identified a number of genes involved in sister chromatid cohesion (SCC) to have synthetic defects with the Origin Recognition Complex defective mutants, orc2 and orc5. These SCC genes include DCC1, CTF4, CTF8 and CTF18, which encode subunits of the alternate RFC complex important for sister chromatid cohesion. While the SGA study found that ORC plays a role in SCC, a conclusion supported by another study,Citation6 it categorically ruled out that the SCC genes, in particular CTF4 and CTF18, play any role in the initiation of DNA replication. By contrast, the Ma study showed convincingly that Ctf1 and Ctf18 are directly involved in DNA replication initiation based on their association with ORC, Cdt1 and MCM proteins, their requirement in S-phase entry, S-phase progression and preRC formation. Furthermore, they showed that Ctf4 plays a role in maintaining the association of Mcm10 and DNA polymerase α.Citation7 Why are there such discrepancies? The diametrically opposite conclusions stem from the use of deletion mutations in SGA versus point mutations in the random mutagenesis study. As it turns out, just like ORC, the SCC proteins are involved in both replication initiation and sister chromatid cohesion and that these two functions are genetically separable. In such synthetic defect analyses, the use of deletion mutations that harbor more than one defect may cloud conclusions.
What is satisfying about the Ma et al. study is that along with the idr-SCC genes, they have also identified a significant number of other previously unknown IDR genes including CDC14 and ADK1. In a separate paper, the same group showed that CDC14 plays an essential role in dephosphorylating multiple initiation proteins to allow preRC formation during the M-G1 transition.Citation8 A more surprising finding is perhaps the direct involvement of ADK1, which encodes an adenylate kinase, in providing ATP for pre-RC assembly.Citation9 This finding may revive an old notion that nucleotide substrates for specific DNA metabolism may be directly channeled by the relevant nucleotide biosynthetic enzymes as suggested for the channeling of hydroxymethyl dCTP for DNA synthesis in the T-even phages.Citation10 There is still much to be learned about the interconnecting processes that weave the cell division cycle using new and old global genetic analyses. An important lesson from the Ma et al. study is that there is plenty of room for old tricks to turn new profits.
References
- Tong AH, et al. Science 2001; 294:2364 - 2368
- Hartwell LH, et al. Genetics 1973; 74:267 - 286
- Maine GT, et al. Genetics 1984; 106:365 - 385
- Ma L, et al. Cell Cycle 2010; 9:4399 - 4410
- Suter B, et al. Genetics 2004; 167:579 - 591
- Shimada K, et al. Cell 2007; 128:85 - 99
- Wang J, et al. Biochem Biophys Res Commun 2010; 395:336 - 341
- Zhai Y, et al. J Cell Sci 2010; 123:3933 - 3943
- Cheng X, et al. J Biol Chem 2010; 285:29974 - 29980
- Flanegan JB, et al. J Biol Chem 1977; 252:3019 - 3027
Histone synthesis is highly regulated in all eukaryotes, and has served as a paradigm for understanding cell cycle regulated-transcription,Citation1 post-transcriptional RNA processing,Citation2 and regulated protein decay.Citation3 Improper histone expression can have deleterious effects on cell-cycle progression,Citation4 chromosome stabilityCitation5 and gene expression.Citation6 Two recent papers show there are many other effects, describing new phenotypes associated with histone overexpression in budding yeast.
In previous experiments, Akash Gunjan and co-workers observed that budding yeast cells lacking checkpoint kinase Rad53 are very sensitive to histone overexpression. This lead to the discovery Rad53 has a novel, non-checkpoint related role in degradation of excess soluble histones.Citation3 Specifically, histone phosphorylation by Rad53 triggers proteosomemediated degradation.Citation7 In a previous issue of Cell Cycle, this laboratory explored multiple ways in which excess histones affect cells. Multiple effects are observed: yeast mutants lacking several different histone modifying enzyme subunits are shown to be sensitive to histone H3 overexperession, suggesting that excess histones might overload modification pathways. H3 overexpression also results in reduced nucleosomal linker length and altered nucleosome positions in vivo. Upon analysis of steady-state mRNA levels, few genes are altered upon overexpression of H3 alone, a somewhat surprising result given the chromatin structural changes documented above. However, over-expression of an H3/H4 gene pair or all four core histones resulted in >200 loci with >2-fold alterations in mRNA levels. Curiously, several clusters of adjacent genes were among these, suggesting that specific chromatin neighborhoods are particularly sensitive to histone density. Perhaps these observations are related to older findings that overexpression of histone pairs but not individual histones causes chromosome loss.Citation5 Finally, the investigators show that excess histones can be crosslinked to RNA in vivo, raising the possibility that excess histones could exert some of their effects via sequestration of RNAs.
Those sorts of observations might lead one to predict that histone overexpression would promote reduced viability or shorter lifespans. However, recent work from Jessica Tyler and co-workersCitation8 suggests just the opposite: histone H3/H4 (but not H2A/H2B) overexpression lengthens the budding yeast replicative life-span. Conversely, replicatively old cells display reduced histone levels. These data suggest that depletion of histones during aging could lead to undesirable access to DNA, perhaps via cryptic transcription; in this view, extra histones would offset this loss.
How can we consolidate these seemingly contradictory observations that histone overexpression can perturb chromosomes yet still extend lifespan? A comprehensive model would suggest that cells are able to cope with minor perturbations caused by histone overexperssion when they are young (as long as the Rad53-mediated decay pathway is intact, so that grossly toxic occlusion of DNA is avoided). In contrast, in old cells the reduced amount of histones reveals the benefit of a fully packaged genome, such that histone overexpression prevents many of the deleterious effects of age-associated histone loss in old cells. In the future, it will be interesting to determine the extent that the phenomena of kinase-mediated destruction of nascent molecules and age-associated histone loss are conserved in other eukaryotes.
References
- Osley MA. Annu Rev Biochem 1991; 60:827 - 861
- Marzluff WF, et al. Curr Opin Cell Biol 2002; 4:692 - 699
- Gunjan A, et al. Cell 2003; 115:537 - 549
- Kim UJ, et al. EMBO J 1988; 7:2211 - 2219
- Meeks-Wagner D, et al. Cell 1986; 44:43 - 52
- Wyrick JJ, et al. Nature 1999; 402:418 - 421
- Singh RK, et al. Nat Cell Biol 2009; 11:925 - 933
- Feser J, et al. Mol Cell 2010; 39:724 - 735
- Singh RK, et al. Cell Cycle 2010; 9:4236 - 444
Acute myeloid leukemia (AML) is a heterogeneous disease in which multiple genetic and epigenetic defects lead to abnormal differentiation and unlimited self-renewal of hematopoietic stem/myeloid progenitor cells.Citation1 AML is a difficult disease to treat, especially in the case of refractory, relapsed and elderly AML patients.Citation2 Prognosis is highly dependent on age at presentation and the underlying molecular defect. Hence, cytogenetics and molecular biology are increasingly being used in diagnosis and to determinate optimum therapy.Citation3 In certain subsets of patients mean survival time and overall cure rate have been greatly improved with patient-tailored chemotherapeutic regimens and drugs targeting the molecular defects driving proliferation/survival of neoplastic cells.Citation3
It has been known for ∼30 years that 1α,25-(OH)2D3 induces growth arrest and monocytic differentiation of cultured AML cells. Even so, 1α,25-(OH)2D3 isn't used therapeutically, largely due to an unacceptable frequency of occurrence of hypercalcemia and a high proportion of non-responding patients.Citation4,Citation5 Analogs with low calcemic capacity have been developed, but none of these have been shown to be clinical useful. Efforts are under way to decrease the likelihood of 1α,25-(OH)2D3-mediated toxicity by increasing the sensitivity of AML cells to 1α,25-(OH)2D3, by co-administration of “differentiation potentiating” agents. Alternatively, specific inhibition of pathways that mediate 1α,25-(OH)2D3 resistance may produce clinically beneficial effects.
Seminal work on 1α,25-(OH)2D3-induced monocytic differentiation of AML cells has emanated from the Studzinski lab (UMD-New Jersey Medical School, Newark, NJ). Recent studies have centred on identifying the mechanisms by which 1α,25-(OH)2D3 activates the p42 ERK, JUN and p38 MAP kinase signaling pathways and unravelling the complex stimulatory and inhibitory interactions between these pathways.Citation6 In particular, Studzinski's group have been at the forefront of research into the use of novel drug combinations as treatments for AML. This research has shown that silibinin, a plant antioxidant, potentiates 1α,25-(OH)2D3-mediated differentiation in some AML cell lines and primary cells by augmenting MAP kinase signalling pathways. Unfortunately, several sub-sets of primary AML cells still remained 1α,25-(OH)2D3-resistant.Citation5 In a previous issue of Cell Cycle, Wang et al.Citation7 shedded new light on resistance by identifying a 1α,25-(OH)2D3- and silibinin-activated signalling pathway that negatively regulates monocytic differentiation in several types of AML cells. The Cot1 proto-oncogene, a member of the MAP kinase kinase kinase (MAP3K) superfamily, and its downstream target the serine threonine kinase ERK5 are key components of this pathway. Cot1 has been reported to be involved in regulating activities of the differentiation promoting p42 ERK, JUN and p38 and the differentiation restraining ERK5 MAP kinase signalling pathways. Earlier this year Studzinski's group demonstrated that expression of Cot1, and its kinase activity, was rapidly increased by 1α,25-(OH)2D3 in HL60 myeloid leukemic cellsCitation7,Citation8 and this disrupts prodifferentiation and growth arresting pathways by interfering with MAPK signalling.Citation9 In other cellular scenario's activation of Cot1 and ERK5 has pro-proliferative effects by down-regulating the expression of, and signalling from, the cell cycle inhibitory protein p27Kip1. Wang et al.Citation7 show that this also occurs in several different subclasses of AML and confirm that Cot1 can act as an oncogene in these cells.
These findings have important implications for the therapy of AML, since small molecule pharmacological inhibitors of Cot1 are available and could be used to remove the ‘molecular brake’ that restrains 1α,25-(OH)2D3-stimulated monocytic differentiation. To this end Wang et al.Citation7 have shown in a previous issue of Cell Cycle that co-administration of silibinin and a Cot1 inhibitor increases the sensitivity of several myeloid leukemic cell lines and primary cells to 1α,25-(OH)2D3. Most importantly they were able show that inhibition of Cot1 was sufficient to induce growth arrest and differentiation in even the most primitive AML cell lines and primary leukemic blasts which are normally differentiation resistant. Cot1 inhibitors were less effective at inducing differentiation of more differentiated AML blasts and cell lines which indicates that high expression of Cot1 may be a major cause of the failure of primitive AML cell types to respond to differentiation inhibitors. The key point to take from these studies is that by thoroughly understanding the molecular mechanisms by which differentiating agents produce their effects, and perhaps more importantly by appreciating why some cell types do not respond, new therapies for hard-to-treat myeloid leukemias will undoubtedly be uncovered.
References
- Vardiman JE, et al. Blood 2009; 114:937 - 951
- Motyckova G, et al. Curr Hematol Malig Report 2010; 5:109 - 117
- Döhner H, et al. Blood 2010; 115:453 - 474
- Gocek E, et al. Leuk Res 2010; 34:649 - 657
- Zhang J, et al. Hematol Oncol 2010; 28:124 - 132
- Hughes PJ, et al. Leuk Res 2010; 34:553 - 565
- Wang X, et al. J Cell Cycle 2010; 9:4542 - 4551
- Wang X, et al. J Steroid Biochem Mol Biol 2010; 121:395 - 398
- Wang X, et al. J Cell Physiol 2010; DOI:JCP-10-0611
Considered the “engines” of cell cycle progression, cyclin-dependent kinases (CDKs) phosphorylate myriad downstream substrates to promote cell growth, replication and division. Tight control of CDK activity results from, among other mechanisms, the opposing actions of inhibitory kinases (Wee1 and Mik1) and activating phosphatases. Cdc25 phosphatases are essential components of this process, reversing inhibitory phosphorylation of CDKs during key cell cycle transitions and peaking in their activity as cell division nears its mitotic end.
Although the three mammalian Cdc25 paralogs—Cdc25A, Cdc25B and Cdc25C—are regulated by multiple mechanisms, degradation of Cdc25A and Cdc25B by the ubiquitin-proteasome system remains a major method of regulation during the cell cycle. Interestingly, Cdc25 genes are considered proto-oncogenes whose excessive activity accelerates proliferation, but the increased levels of Cdc25A and Cdc25B observed in tumors do not generally occur as a consequence of amplification or mutations that lead to overt overexpression. Instead, elevated Cdc25 levels result from their stabilization and persistence during cell cycle periods when they should be absent or degraded.Citation1
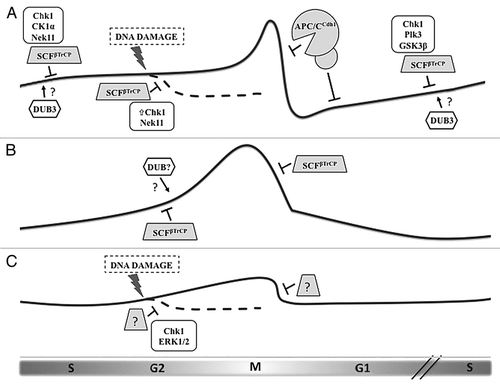
The functional characteristics that distinguish the Cdc25 paralogs have been increasingly investigated. While the mouse knockout model of Cdc25A displays embryonic lethality, deletion of either Cdc25B or Cdc25C imparts little phenotypic outcome at both the organismal and cellular levels.Citation2 In fact, cells derived from mice lacking both Cdc25B and Cdc25C do not show significant defects in their cell cycles or DNA damage responses, with these normal phenotypes perhaps indicating redundancy in their functions and substrates. In contrast to these loss-of-function studies, examining the aberrant persistence of these proteins may reveal more about their particular roles and elucidate more subtle mechanisms of their regulation. Studies investigating the specific conditions for Cdc25A degradation mediated by the Anaphase Promoting Complex/Cyclosome (APC/CCdh1) or the SCFβTrCP ubiquitin ligases have led to a greater understanding of the delicate balance of Cdc25A levels across the cell cycleCitation3 (). Now, by confirming and extending the long-postulated notion that Cdc25B, like Cdc25A, is degraded by the ubiquitin-proteasome system via the F-box protein βTrCP,Citation4 the work of Thomas et al. in a previous issue of Cell Cycle furthered this discussion of distinct roles within the Cdc25 family.Citation5
In their report, the authors investigate the unique features of Cdc25B activity during mitosis, showing βTrCP-dependent degradation at the metphase-anaphase transition—in surprising contrast to the nearly simultaneous removal of Cdc25A through APC/CCdh1—and raise intriguing questions about novel mechanisms regulating βTrCP-mediated ubiquitylation. In S phase, when the activity of CDKs must be maintained at levels low enough to be permissive for DNA replication, Cdc25A levels are moderated by βTrCP. Following S-phase, CDK activity rises, inhibiting replication (thereby preventing reduplication) and promoting mitotic events.Citation3 In response to DNA damage in S and G2, βTrCP-dependent degradation of Cdc25A is enhanced. These interphase and DNA damage-responsive interactions between Cdc25A and βTrCP require phosphorylation by a number of kinases—one priming (Chk1),Citation3 and others processive (CKIα, GSK3β and Nek11),Citation6,Citation7 with binding dependent upon the final phosphorylation within the protein's βTrCP degron, an inverted variant of the classical DSGxxS motif (STDSG).
The many characterized substrates of βTrCP share some version of this DSGxxS phosphodegron, sometimes with phosphomimetic substitution, but Thomas et al. instead show that Cdc25B uniquely lacks any phosphorylable residue in its degron sequence (DDGFVD). Since regulation of the timely recognition of substrates by βTrCP famously follows a phosphorylation within this motif, how might Cdc25B ubiquitylation be controlled?
The authors speculate that regulation of Cdc25B degradation in the absence of a phosphorylable degron may be due to modification of βTrCP itself, or it may represent a kind of competition for βTrCP, in which occupation of its substrate recognition site by phosphorylated substrates (which are likely to bind βTrCP with high affinity) allows Cdc25B to accumulate, but permits its degradation at the end of mitosis after other substrates have been eliminated. The abundance of βTrCP in the cell and its diverse substrate targets argue that perhaps another mechanism might exist. One way to counteract a ‘constitutively active’ degron would be to oppose ubiquitylation in a regulated manner, namely using a deubiquitylating enzyme (DUB). Thus, it is possible that a yet undiscovered DUB stabilizes Cdc25B at the G2/M transition. For example, recent evidence indicates that Dub3 (also called USP17) positively regulates Cdc25A levels and has been found to be elevated in human breast cancer cell lines in which Cdc25A is stabilized.Citation8 Dub3 may therefore contribute to tumorigenesis by inappropriately stabilizing Cdc25A and, conceivably, other substrates.
High expression of exogenous Cdc25B is known to induce mitotic catastrophe, and overexpression of Cdc25B in S phase results in centrosome overduplication.Citation9 In their report, Thomas et al. show that a stabilized Cdc25B mutant (unable to bind βTrCP) yields discrete cellular consequences, including a delay in mitotic exit, diverse chromosomal and spindle defects and the appearance of micronuclei that accrue in subsequent mitoses. Therefore, the regulation of Cdc25B turnover in the absence of a phosphorylable degron is a subject of interest for future studies of Cdc25 phosphatase activity during the cell cycle, and exploration of alternative regulatory mechanisms may provide a greater overall understanding of regulated proteolysis.
Figures and Tables
Figure 1 Regulation of the three CDC25 paralogs during the cell cycle by ubiquitin-mediated degradation. (A) Cdc25A protein levels begin to rise in late G1 and remain modest during S phase and early G2 by the action of the ubiquitin ligase SCFβTrCP, whose recognition of Cdc25A depends upon phosphorylation by a number of kinases. DNA damage induction of Chk1 activity increases the priming phosphorylation of Cdc25A, leading to greater βTrCP-induced degradation. At the end of mitosis and through G1, Cdc25A is eliminated via the APC/CCdh1 ubiquitin ligase. Dub3 has been shown to stabilize Cdc25A by deubiquitylation, although the precise timing and conditions for its activity remain unknown. (B) Cdc25B levels are also moderated by βTrCP, but in a phosphorylation-independent manner. Rapid destruction of Cdc25B at the metaphase-anaphase transition is mediated by βTrCP. Since the degron of Cdc25B mimics constitutive phosphorylation, we hypothesize that another mechanism counteracts βTrCP-mediated ubiquitylation during its rise in G2, possibly the activity of a deubiquitylating enzyme (DUB). (C) Cdc25C is the least-studied protein of the family, and its regulation is determined mostly by phosphorylation events that regulate its phosphatase activity and localization. Evidence exists that Cdc25C is ubiquitylated and degraded following certain forms of G2 arrest, but, although this event requires Chk1 and ERK1/2, the contributing ubiquitin ligase is unknown.
References
- Löffler H, et al. Oncogene 2003; 22:8063 - 8071
- Guardavaccaro, et al. Mol Cell 2006; 22:1 - 4
- Frescas D, et al. Nat Rev Cancer 2008; 8:438 - 449
- Kanemori Y, et al. Proc Natl Acad Sci USA 2005; 102:6279 - 6284
- Thomas Y, et al. Cell Cycle 2010; 9:4338 - 4350
- Melixetian M, et al. Nat Cell Biol 2009; 11:1247 - 1253
- Honaker Y, et al. Oncogene 2010; 29:3324 - 3334
- Pereg Y, et al. Nat Cell Biol 2010; 12:400 - 4006
- Boutros R, et al. Cancer Res 2007; 67:11557 - 11564
Each time a cell divides, the whole genome must be duplicated and the two copies faithfully separated between the mother and the daughter cells. This is achieved by a complex mechanism that involves maintaining the newly replicated and the old DNA molecules (the two sister chromatids) attached while building the separating machinery. Sister chromatid cohesion (SCC) is being actively investigated in yeast.Citation1 The cohesin complex, composed of the Smc1, Smc3, Scc1/Mcd1 and Scc3 subunits, forms a ring that is essential for cohesion. Two additional protein heteroduplexes are required: the Scc2-Scc4 and the Pds5-Rad61/Wpl1 complexes. Finally, the Ctf7/Eco1 acetyltransferase plays an important regulatory role by acetylating Smc3 during S phase; this acetylation is essential to establish cohesion, by a mechanism that is being actively studied.Citation2–Citation4
In the last years, a new family of proteins has been found to affect SCC: the Replication Factor C (RFC) family. The canonical RFC complex plays an essential role in loading the processivity clamp PCNA onto DNA. RFC is composed of four small (Rfc2-5) and a large subunit (Rfc1). Three additional RFC-like factors exist in yeast; in these complexes the large subunit is replaced by the Rfc1-like proteins Rad24, Ctf18 or Elg1.Citation5 Whereas Ctf18 and Elg1 have been shown to interact with PCNA, Rad24 loads the PCNA-like clamp 9-1-1 that plays an important role in checkpoint induction. Mutations in either Elg1 or Ctf18 cause defects in SCC.Citation6,Citation7
Previous results by the Skibbens lab have shown that a deletion of the ELG1 gene can partially rescue the lethal effects of the ctf7_eco1 mutation, whereas deletion of CTF18, when combined with that mutation, causes lethality.Citation8 In a previous issue of Cell Cycle,Citation9 Maradeo and Skibbens investigated whether mutations in one of the shared small subunits, Rfc5, may also affect SCC. Being part of both the Elg1 and Ctf18 complexes, it is unclear a priori what phenotype is to be expected.
A double mutant rfc5-1 eco1 exhibited viability at temperatures at which the single eco1 mutant is inviable, demonstrating that the mutation at RFC5 has an effect similar to deleting ELG1. Significantly, additional mutation of ELG1 did not have a further effect, confirming that in these crosses the Rfc5 defect exerts its phenotype through Elg1, and not through Ctf18. Double mutants rfc5-1 scc1 or rfc5-1 smc3-5 show a reduction in viability at low temperatures, at which the single mutants are viable, again showing an elg1-like effect. Similar results were obtained when combining rfc5-1 with Scc2. Notably, in all these crosses, the phenotype of the rfc5-1 mutant was milder than that obtained in the absence of Elg1. It is unclear at the moment if this is due to the fact that the former is a hypomorphic allele, whereas the latter completely lack protein, or whether these results indicate that Elg1 may have Rfc5-independent functions.
The striking effects observed for a mutation in a subunit that is shared by four different complexes are surprising. The authors propose several mechanisms that may explain the role played by Rfc5: It may directly interact with cohesins or cohesin-deposition factors, or may play a role as sensor to allow recruitment of a specific RFC complex, depending on the cell's needs. Alternatives include the possibility that sub-complexes containing only part of the RFC subunits may play additional roles. Such complexes have been shown to be active in vitro.
This paper contributes to the large number of observations recently published, which require fresh models about the mechanism that establishes cohesion. It is becoming increasingly clear that such a mechanism occurs in multiple, regulated steps, which are tightly coordinated with the passage of the DNA polymerase complex during DNA replication, perhaps by PCNA modification.Citation7 Further genetic and biochemical evidence is necessary to unravel the complexity of this important step in the cell cycle.
References
- Skibbens RV. Curr Biol 2009; 19:R1126 - R1132
- Beckouet F, et al. Mol Cell 2010; 39:689 - 699
- Borges V, et al. Mol Cell 2010; 39:677 - 688
- Xiong B, et al. Curr Biol 2010; 20:1660 - 1665
- Aroya SB, et al. DNA Repair 2005; 4:409 - 417
- Parnas O, et al. PLoS One 2009; 4:e5497
- Parnas O, et al. EMBO J 2010; 29:2611 - 2622
- Maradeo ME, et al. PLoS One 2009; 4:e4707
- Maradeo ME, et al. Cell Cycle 2010; 9:4370 - 4378