Abstract
Emerging research shows that the packaging of mitochondrial DNA (mtDNA) into protein-DNA assemblies called nucleoids confers higher-order organization to the mitochondrial genome. Studies of nucleoid composition, structure, and dynamics reveal the mitochondrial nucleoid to be tightly regulated in its genetic autonomy, macromolecular organization, and distribution throughout the cell. Our recent research shows that mitochondrial nucleoids are self-contained genetic entities that do not exchange mtDNAs with each other frequently. This suggests that the genetic composition of a cell’s nucleoids will be the key determinant of the cell’s mtDNA dynamics, and provides a mechanistic basis for therapeutic methods to rescue dysfunction due to mutations in mtDNA.
The human mitochondrial genome is a 16,569 bp circular DNA, essential for mitochondrial ATP production, which is present in ∼1000 copies per cell. MtDNA is packaged into macromolecular assemblies called nucleoids, which consist of mtDNA and its associated proteins. While it has been widely supposed that mtDNA is largely ‘naked’ and therefore exposed to reactive oxygen species and other insults, current research suggests instead that nucleoids tightly regulate the genetic content, packaging and intracellular distribution of mtDNA. Moreover, the genetic autonomy of mitochondrial nucleoids provides a molecular mechanism to explain contradictory observations of mtDNA maintenance and inheritance, with important implications for therapeutic approaches to mitochondrial diseases.
Increasingly, the mitochondrial nucleoid is revealed to be a genetically autonomous, highly regulated assembly for the maintenance and propagation of mtDNA. The mitochondrial transcription factor TFAM associates with mtDNA nucleoids and regulates mtDNA copy number.Citation1 TFAM has been estimated to exist at ∼900 copies per mtDNA molecule, consistent with its presence in excess of mtDNA copy number due to its vital role in maintaining mtDNA integrity.Citation2 TFAM has a significant capacity for bending and packaging mtDNA into nucleoid-like multicopy structures; further, the affinity of TFAM for DNA is enhanced by the presence of bound TFAM,Citation3 suggesting that, in vivo, there is very little mtDNA which is not packaged and complexed with protein, even during replication and transcription. Other nucleoid-associated proteins include prohibitin,Citation4 mitochondrial single strand binding protein, mitochondrial polymerase γ, and the Twinkle helicase.Citation5 It is likely that nucleoids associate with the mitochondrial inner membrane via protein-protein interaction,Citation5 indicating that nucleoids are ‘tethered’ to the inner membrane,Citation6 although the precise interactions are unclear.
Within the mitochondrial network, mitochondrial nucleoids appear as punctae distributed along the length of mitochondrial filaments;Citation7,Citation8 when mitochondria transition to a fragmented morphology, each mitochondrion will contain at least one nucleoid.Citation9 This suggests that nucleoids are compartmentalized within the mitochondrion,Citation10 although how mitochondria ‘know’ where to position nucleoids is completely unknown. Our recent work has revealed that mitochondrial nucleoids regulate their genetic content tightly: in a fused-cell system, two heterologous nucleoid populations remained independent, rather than stably exchanging mtDNAs, when visualized by two-color fluorescence in situ hybridization. Further, the two heterologous mtDNAs did not recombine with each other.Citation11 This is consistent with faithful nucleoid inheritance, as first proposed by Jacobs et al.,Citation12 in which nucleoids replicate their genetic content exactly to produce identical daughter nucleoids. This genetic autonomy, combined with the packaging of mtDNA by nucleoid-associated proteins and the regular arraying of nucleoids within the mitochondrion, suggests that the mitochondrial nucleoid is a highly regulated, self-contained genetic entity, ensuring that mtDNA is distributed efficiently throughout the mitochondrial network.
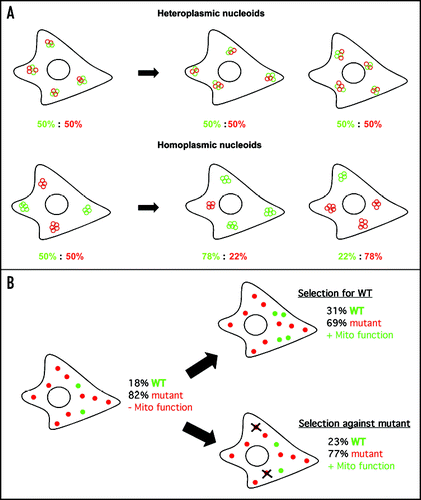
The genetic autonomy of mitochondrial nucleoids represents an underlying mechanistic basis for often-confusing patterns of mtDNA inheritance and propagation, as well as providing a potential molecular “in” for the rescue of mitochondrial dysfunction. Mutations of mtDNA, both point mutations and large-scale deletions (Δ-mtDNAs), cause a host of systemic and tissue-specific diseases,Citation13 and correlate with, if not actually cause, common neurodegenerative diseases, such as Parkinson disease and aging.Citation14,Citation15 Frequently, wild-type (WT) and mutant mtDNAs co-exist within the same tissue, cell and organelle, a situation called heteroplasmy. MtDNA heteroplasmy displays contradictory and confusing patterns of inheritance, undergoing random segregation, stable heteroplasmy, or rapid selection in different situations and cell types.Citation16–Citation18 In addition, although WT and mutant mtDNAs often exist in close proximity, recombination between mtDNAs is extremely rare. Faithful nucleoid inheritance suggests that the mtDNA composition of a cell's nucleoids will be the key determinant of cellular heteroplasmy over time. If all nucleoids have approximately the same mtDNA makeup, the cell's overall heteroplasmy will reflect this in a stable manner (, top). Conversely, if two mtDNA variants are maintained in different nucleoid populations, the overall heteroplasmy will be subject to segregation, depending on selective pressure and random genetic drift (, bottom).
Importantly, cells can often tolerate high levels of mutant mtDNA, until the threshold of mutation load is exceeded and mitochondrial function is compromised. This threshold varies with mutation type, but cells typically can withstand mutation loads of up to 80–90% mutant mtDNA by compensating with the remaining minority population of WT mtDNAs (reviewed in ref. Citation13). We had shown previously that a respiratory-deficient cybrid cell line carrying 13% WT mtDNA and 87% Δ-mtDNA (above threshold), when treated with ketogenic media to activate mitochondrial ATP production, shifted its heteroplasmy to 22% WT/78% Δ-mtDNA (below threshold), resulting in a dramatic recovery of mitochondrial protein synthesis and WT-like mitochondrial morphology.Citation19 Due to the threshold effect of mtDNA heteroplasmy, this small increase in the proportion of WT mtDNAs effectively breached the threshold of mutation load, thereby rescuing mitochondrial function. If WT and mutant mtDNAs exist in separate nucleoid populations, then the two discrete nucleoid populations create an intracellular environment with a dynamic mtDNA heteroplasmy that is capable of being shifted rapidly. Therefore, it should be possible to select for WT mtDNA-containing nucleoids without concomitant amplification of mutant mtDNA (i.e., by positive selection), or, conversely, mutant mtDNA may be eliminated without losing WT mtDNA content, as by autophagy (). A great advantage of these approaches is that they involve metabolic adjustment of innate cellular processes, rather than harsh pharmacological treatments or viral introduction of exogenous transgenes, which may have unforeseen toxic side effects. Further, the ketogenic diet is already an existing therapeutic method; it will be interesting to see whether it can be applied to the rescue of mitochondrial dysfunction.
Figures and Tables
Figure 1 (A) The mtDNA content of a cell's nucleoids will determine cellular heteroplasmy. In a cell containing two mtDNA variants (green and red circles), which are packaged into nucleoids containing 4–5 copies of mtDNA, the mtDNA content of the individual nucleoids will determine the heteroplasmic variability of the cell. If the individual nucleoids are heteroplasmic (top), each carrying both mtDNA variants in roughly equal proportion, the cell's overall heteroplasmy will be 50% green: 50% red. If, however, the individual nucleoids are homoplasmic (i.e., carrying only one type of mtDNA, bottom), the cellular heteroplasmy of the cell and mitotic daughter cells may vary widely. The homoplasmic nucleoid model as shown assumes random genetic drift of the two variants; however, such cells would also be highly susceptible to both selective pressure and replicative advantage. (B) Small shifts in heteroplasmy can rescue mitochondrial function. In heteroplasmic cells carrying high levels mutant mtDNAs with a minority of WT mtDNA above the mutation load threshold, either a small increase in WT mtDNA or a small decrease in mutant mtDNA may have the effect of restoring mitochondrial function. Each circle represents one nucleoid carrying 5 copies of mtDNA (green = WT, red = mutant). If the heteroplasmic cell shown on the left has 9 nucleoids carrying mutant mtDNA and 2 carrying WT mtDNA for a total heteroplasmy of 18% WT: 82% mutant, the cell will be above the phenotypic threshold, resulting in mitochondrial functional deficiency. Selection for WT, adding only two additional WT nucleoids, will push the proportion of WT mtDNA to 31%, pushing the net heteroplasmy below the phenotypic threshold, rescuing mitochondrial function. Selection against mutant mtDNA, removing two nucleoids, pushes the net proportion of WT to 23%, below threshold and also rescuing mitochondrial function.
Acknowledgements
This research was supported by grants from the National Institutes of Health (HD83062, NS11766 and AG08702 to Eric A. Schon), the Marriott Foundation, Edison Pharmaceuticals, the American Health Assistance Foundation, and the Muscular Dystrophy Association (MDA3869 to Robert W. Gilkerson). The authors declare that they have no competing financial interests.
Addendum to:
References
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 2004; 13:935 - 944
- Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, Takio K, Hamasaki N, Kang D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res 2003; 31:1640 - 1645
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell 2007; 18:3225 - 3236
- Kasashima K, Sumitani M, Satoh M, Endo H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp Cell Res 2008; 314:988 - 996
- Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem 2006; 281:25791 - 25802
- Albring M, Griffith J, Attardi G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc Natl Acad Sci USA 1977; 74:1348 - 1352
- Legros FMF, Frachon P, Lombes A, Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci 2004; 117:2653 - 2662
- Iborra FJ, Kimura H, Cook PR. The functional organization of mitochondrial genomes in human cells. BMC Biol 2004; 2:9
- Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA. Cell cycle dependent morphology changes and associated mitochondrial DNA. Mitochondrion 2002; 1:425 - 435
- Capaldi RA, Aggeler R, Gilkerson R, Hanson G, Knowles M, Marcus A, Margineantu D, Marusich M, Murray J, Oglesbee D, Remington SJ, Rossignol R. A replicating module as the unit of mitochondrial structure and functioning. Biochim Biophys Acta 2002; 1555:192 - 195
- Gilkerson RW, Schon EA, Hernandez E, Davidson MM. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J Cell Biol 2008; 181:1117 - 1128
- Jacobs HT, Lehtinen SK, Spelbrink JN. No sex please, we're mitochondria: a hypothesis on the somatic unit of inheritance of mammalian mtDNA. Bioessays 2000; 22:564 - 572
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003; 348:2656 - 2668
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006; 38:515 - 517
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 2006; 38:518 - 520
- Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 1996; 14:146 - 151
- Yoneda M, Chomyn A, Martinuzzi A, Hurko O, Attardi G. Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc Natl Acad Sci USA 1992; 89:11164 - 11168
- Rajasimha HK, Chinnery PF, Samuels DC. Selection against pathogenic mtDNA mutations in a stem cell population leads to the loss of the 3243A→G mutation in blood. Am J Hum Genet 2008; 82:333 - 343
- Santra S, Gilkerson R, Davidson M, Schon EA. Ketogenic treatment reduces the proportion of mutated mitochondrial DNAs in cells harboring mtDNA deletions. Ann Neurol 2004; 56:662 - 669