Abstract
We recently described Megavirus chilensis, a giant virus isolated off the coast of Chile, also replicating in fresh water acanthamoeba. Its 1,259,197-bp genome encodes 1,120 proteins and is the largest known viral genome. Megavirus and its closest relative Mimivirus only share 594 orthologous genes, themselves sharing only 50% of identical residues in average. Despite this divergence, comparable to the maximal divergence exhibited by bacteria within the same division (e.g., gamma proteobacteria), Megavirus retained all of the genomic features unique to Mimivirus, in particular its genes encoding key-elements of the translation apparatus, a trademark of cellular organisms. Besides homologs to the four aminoacyl-tRNA synthetases (aaRS) encoded by Mimivirus, Megavirus added three additional ones, raising the total of known virus-encoded aaRS to seven: IleRS, TrpRS, AsnRS, ArgRS, CysRS, MetRS, TyrRS. This finding strongly suggests that large DNA viruses derived from an ancestral cellular genome by reductive evolution. The nature of this cellular ancestor remains hotly debated.
The era of giant viruses (Giruses) started in 2003 with the discovery and genome sequencing of the first of them, called MimivirusCitation1,Citation2 (for Microbe Mimicking virus), originally mistaken for a new amoeba-resisting intracellular parasitic bacteria (reviewed in ref Citation3). The main common features of Giruses are their large pseudo-icosahedral capsids (with diameter > 400 nm), most often enclosed in a thick (≈100 nm) layer of fibers, a large double-stranded DNA genome (≈a million bp) and a large gene content (approximately 1000 genes). Few have been characterized in details, but all the Mimivirus-like isolates replicate from large circular intracytoplasmic virion factories, as previously described for poxviruses.Citation4 For Mimivirus and its nearly identical relative Mamavirus, these cell-like virion factories can themselves be infected by a new type of satellite virus called “virophage”Citation4,Citation5. The last, but not the least of the unique puzzling features of Mimivirus is the presence of many genes coding for central elements of the translation apparatus, until now thought to be an absolute trademark of cellular organisms. Mimivirus, for instance, encodes 4 aminoacyltRNA synthetases (for Arg, Cys, Met, and Tyr). This finding, among others, triggered a heated debate on the evolutionary origin of these viruses,Citation5-Citation8 opposing the traditionalists school viewing them as tremendously efficient pickpockets of cellular genes,Citation9 up to the most extreme views of those claiming them as evidence of a fourth domain of life,Citation10 or as a dismissal of darwinism.Citation11 Our discovery and genomic sequencing of Megavirus chilensis, a girus with an even larger capsid, genome size and gene content,Citation12 first demonstrated that the oddities observed in Mimivirus and its closest relatives are not anecdotal, but intrinsic features shared by more distant giant viruses, and inherited from their common ancestor. Among these, the most significant is the presence of the same four aminoacyl-tRNA synthetases found in Mimivirus, together with three more, including the IleRS, also found in the third known largest virus infecting the heterotrophic marine protozoan Cafeteria roenberbengis.Citation13 These findings strongly support a scenario whereby these large DNA viruses all evolved from an ancestral cellular genome by reductive evolution.
A New Rapid Genome Sequencing Strategy for Microorganisms
Only 15 mo separated the water sampling in Chile (mid-April 2010) from the submission of the complete annotated Megavirus genome sequence to GenBank (mid-July 2011). This was made possible by the combination of two short read sequencing approaches that nicely complement each other: the Roche 454-titanium and the Illumina Hiseq techniques. Nowadays, most of de novo microbial whole genome sequences are produced using the 454 platform. This technology provides long-enough sequence reads to rapidly obtain few contigs of reasonable lengths. Unfortunately the technology is also error prone within homopolymeric regions, ultimately leading to frequent frame shift errors when annotating protein coding genes. Therefore finishing may be quite cumbersome, involving numerous PCR, or worse conventional library-based Sanger sequencing, in order to correct for sequencing errors and close gaps. This is obviously highly labor intensive.
Megavirus chilensis’ genome is highly AT-rich (75% A+T) which raises the probability of finding A or T homopolymers. Indeed, stretches of 6 As or more, or 6 Ts or more, occur 6,186 times in the Megavirus genome, making it highly susceptible to this type of sequencing errors. We circumvented the tedious individual finishing experiments by using the following approach. After an initial assembly using 278,663 454-titanium fragment reads, we re-sequenced the same DNA using one tenth of a lane of Illumina Hiseq and mapped the 42,288,396 resulting paired-end reads to it (approximately a 2000 X coverage!). Although this may appear outrageous to the traditional genome sequencers, this ultra-high-coverage allowed the correction of all sequencing errors (see ) at a very low cost both in terms of direct cost (about 2000 €) and technical labor. We corrected 300 errors in the raw 454-derived Megavirus genome sequence at once.
Figure 1. Effect of sequence correction on read coverage. Visualization of read coverage (blue histogram) resulting from the mapping of Illumina high-throughput sequences at the same genomic locus, prior (A) and following (B) sequence correction. In this example, the deletion of an adenine in an 8-fold A-homopolymer resulted in an increase of coverage where it initially formed a steep drop, and a lengthening of the overlapping ORF (red arrows).
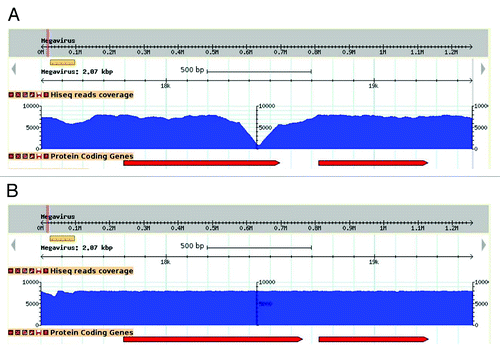
More generally, we think that the approach combining these two NGS platforms should become standard for sequencing microbial genomes, up to 10 Mb in length. In the near future one can expect an even better approach with the combination of the ultra-high coverage short reads (e.g., Illumina Hiseq) with the PacBio much longer reads.Citation14 The latter technology has the capacity to produce sequence reads of up to 10 kb, but remained plagued by much too high an error rate when tentatively applied on the Megavirus genome.
Estimating the gene content of the Megaviridae ancestor
The analysis of the Megavirus gene content revealed 258 proteins with no homolog in the Mimivirus genome.Citation12 Among them 214 (83%) have no significant (E < 10−5) sequence similarity in the public database, and only 34 of them could be associated to a predicted function (). Symmetrically, among the 186 Mimivirus genes without homologs in Megavirus, 149 (80%) are not similar to any other ORF. These large fractions of “ORFans” in the non-overlapping gene contents of Megavirus and Mimivirus definitely rules out that their difference could results from horizontal transfers of genes from cellular organisms (for which the fraction of ORFans would be much lower), or from viruses belonging to a known family. More likely, the difference in gene content is the result of the loss of different genes along the lineage leading to Mimivirus or Megavirus. According to our view, except for some rare cases of lateral gene transfers, most of the 258 Megavirus genes with no homolog in Mimivirus, or conversely of the 186 Mimivirus genes without counterpart in Megavirus, were part of the genome of their common ancestor. Adding the 594 orthologous genes shared by Mimivirus and Megavirus, this predicts an ancestral genome coding for at least 594 + 258 + 186 = 1038 proteins. The actual figure could be higher, if one takes into account the ancestral genes that may have been lost along both the Mimivirus and Megavirus lineages. Our interpretation is at the opposite of the “core” gene subset concept, whereby the ancestral gene pool would solely correspond to the intersection of the Mimivirus and Megavirus gene content (e.g., 594 genes). Our lineage-specific gene loss scenario, predicts that some of the Megavirus gene absent from Mimivirus, could still be found in more distant relatives, such as the Cafeteria roenbergensis virus (CroV). This is actually the case, since eight of the genes (highlighted in gray) listed in do have a homolog in CroV. Equally, four Mimivirus genes not found in Megavirus (R80, R103, R519 and R771) are nevertheless present in CroV. A perfect example of lineage specific loss at work is given by the pattern of presence/absence of the DNA photolyase, an enzyme that repair DNA damage caused by exposure to UV light using the energy of visible light. CroV possesses two intact photolyase genes: crov115 and crov149. Mimivirus only exhibits the remnant of the ortholog to crov149, fragmented into several small ORFs (R852 to R855) flanking an inserted transposase (R854). Finally, Megavirus exhibits one intact photolyase gene (mg779) orthologous to the crov149 gene, while the ortholog to the crov115 genes is now split by a transposase (mg401) into two ORFs: mg400 and mg402. The most parsimonious explanation of this pattern is the presence of two intact photolyase genes in the common ancestor of CroV, Mimivirus and Megavirus, at the moment the sole fully sequenced members of the Megaviridae family.Citation12
Table 1. Megavirus private annotated ORFs
Classifying the Megaviridae: The Fourth Domain of Life Controversy
The feasibility of deep phylogenetic tree reconstruction from viral sequences is a controversial issue. The traditional view, mostly inherited from the study of bacteriophages, considers that (1) it is almost impossible due to the rapid evolutionary rate of viruses, (2) it is non-informative because most of viral sequences are continuously horizontally transferred from cellular (in particular hosts’) genomes. If these criticisms are in part valid within the world of prokaryotes, the situation appears to be not so hopeless for eukaryotic viruses, and especially for large double stranded DNA viruses. For instance, genes specific to a virus family fail to exhibit an accelerated evolutionary rate, compared with genes with more broadly shared homologs.Citation15 Thus the lack of similarity exhibited by a majority of viral genes to cellular counterparts might not be due to their rapid divergence, but more simply to the fact that they have a very ancient origin, within or outside the eukaryotic domain. Also at odds with the traditional view, some studies argue against frequent genetic transfers to large DNA viruses from their modern hosts. The large genome sizes of these viruses are not simply explained by an increased propensity to acquire foreign genes.Citation16
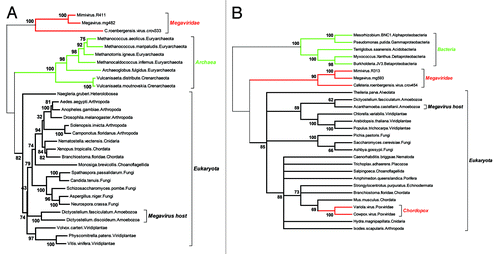
The notion of essential (“core”) genes, already controversial for free leaving organisms, as it depends on the richness of the medium they are grown in, is even more disputable for intracellular parasites and specially viruses, that can easily rely on host-provided biochemical and cellular functions. The ratchet-like process of lineage-specific gene loss is thus expected to shrink the subset of “core” genes common to all (DNA) viruses, as well as of those shared with cellular genomes. The absence of even a single gene both sufficiently universal and sufficiently conserved to serve as a reliable basis for the building of a Tree of Life encompassing all cellular organisms and viruses, is bound to lead to endless controversies.Citation6 Fortunately, some of the Girus genes are conserved enough across two of the three cellular domains, to be the basis of reliable phylogenetic reconstructions. In we used two highly conserved genes, both of them central to the process of DNA replication: the large subunit of the ribonucleoside diphosphate reductase (the enzymatic bridge between RNA and DNA), and the large replication factor C (also known as the DNA replication “clamp loader”). As for previously published phylogenies build with different genes (such as the three novel aaRS found in Megavirus),Citation12 these reconstructions correspond to a tree topology positioning the origin of giant viruses either at the very root of the eukaryotic domain, or downright outside of it. At this point, interpreting such a topology as suggesting a fourth domain vs. a very early split from within the eukaryotic domain remains a matter of personal belief and/or taste for media exposure.
Figure 2. Two reliable phylogenetic reconstructions positionning the Megavirus in a partial Tree of Life. As the quality of the multiple alignment is essential to the reliability of the derived phylogeny, we only included the most similar proteins sequences of each clade in the analyses. (A) Positionning of the three closest Megavirus relatives using the largest clamp loader subunits. The multiple alignment (default options) and tree reconstruction (neighbor joining on 312 ungapped position, JTT substitution model) was performed using the on the MAFFT server (mafft.cbrc.jp/alignment/server/). The highly divergent bacterial homologs were not included, to preserve the quality of the multiple alignment. The deepest bootstrap values indicate the total lack of affinity of the Megaviridae sequences with both the archaeal and eukaryotic domains. (B) Positionning of the three closest Megavirus relatives using their largest ribonucleoside diphosphate reductase subunits. The multiple alignment (default options) and tree reconstruction (neighbor joining on 735 ungapped position, JTT substitution model) was performed as above. This time, the highly divergent archaeal homologs were not included, to preserve the quality of the multiple alignment. The deepest bootstrap values indicate a total lack of affinity of the Megaviridae with the bacterial and eukaryotic domains. In contrast, the acquisition of the vertebrate gene by the chordopoxviruses is showing very clearly, serving as an internal control that virus genes acquired by lateral transfer are indeed detectable. Amoebozoa sequences are indicated in A) and B) to emphasize that the Megavirus/Mimivirus genes do not cluster with their host’s homologs.
Acknowledgments
This work was supported by the Centre National de la Recherche Scientifique, Agence Nationale de la Recherche grant ANRBLAN08–0089, and a fellowship from the Direction Générale de l’Armement (to D.A.). Megavirus chiliensis was isolated from a sampling campaign sponsored by the ASSEMBLE initiative (European Commission’s seventh framework program), grant # 227799.
References
- La Scola B, Audic S, Robert C, Jungang L, de Lamballerie X, Drancourt M, et al. A giant virus in amoebae. Science 2003; 299:2033; http://dx.doi.org/10.1126/science.1081867; PMID: 12663918
- Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H, et al. The 1.2-megabase genome sequence of Mimivirus. Science 2004; 306:1344 - 50; http://dx.doi.org/10.1126/science.1101485; PMID: 15486256
- Raoult D, La Scola B, Birtles R. The discovery and characterization of Mimivirus, the largest known virus and putative pneumonia agent. Clin Infect Dis 2007; 45:95 - 102; http://dx.doi.org/10.1086/518608; PMID: 17554709
- Claverie JM, Abergel C. Mimivirus and its virophage. Annu Rev Genet 2009; 43:49 - 66; http://dx.doi.org/10.1146/annurev-genet-102108-134255; PMID: 19653859
- La Scola B, Desnues C, Pagnier I, Robert C, Barrassi L, Fournous G, et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008; 455:100 - 4; http://dx.doi.org/10.1038/nature07218; PMID: 18690211
- Moreira D, López-García P. Ten reasons to exclude viruses from the tree of life. Nat Rev Microbiol 2009; 7:306 - 11; PMID: 19270719
- Claverie JM, Ogata H. Ten good reasons not to exclude giruses from the evolutionary picture. Nat Rev Microbiol 2009; 7:615; http://dx.doi.org/10.1038/nrmicro2108-c3; PMID: 19561626
- Claverie JM, Abergel C. Mimivirus: the emerging paradox of quasi-autonomous viruses. Trends Genet 2010; 26:431 - 7; http://dx.doi.org/10.1016/j.tig.2010.07.003; PMID: 20696492
- Moreira D, Brochier-Armanet C. Giant viruses, giant chimeras: the multiple evolutionary histories of Mimivirus genes. BMC Evol Biol 2008; 8:12; http://dx.doi.org/10.1186/1471-2148-8-12; PMID: 18205905
- Boyer M, Madoui MA, Gimenez G, La Scola B, Raoult D. Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4 domain of life including giant viruses. PLoS ONE 2010; 5:e15530; http://dx.doi.org/10.1371/journal.pone.0015530; PMID: 21151962
- Raoult D. Giant viruses from amoeba in a post-Darwinist viral world. Intervirology 2010; 53:251 - 3; http://dx.doi.org/10.1159/000312909; PMID: 20551676
- Arslan D, Legendre M, Seltzer V, Abergel C, Claverie JM. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc Natl Acad Sci USA 2011; 108:17486 - 91; http://dx.doi.org/10.1073/pnas.1110889108; PMID: 21987820
- Fischer MG, Allen MJ, Wilson WH, Suttle CA. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc Natl Acad Sci USA 2010; 107:19508 - 13; http://dx.doi.org/10.1073/pnas.1007615107; PMID: 20974979
- Korlach J, Bjornson KP, Chaudhuri BP, Cicero RL, Flusberg BA, Gray JJ, et al. Real-time DNA sequencing from single polymerase molecules. Methods Enzymol 2010; 472:431 - 55; http://dx.doi.org/10.1016/S0076-6879(10)72001-2; PMID: 20580975
- Ogata H, Claverie JM. Unique genes in giant viruses: regular substitution pattern and anomalously short size. Genome Res 2007; 17:1353 - 61; http://dx.doi.org/10.1101/gr.6358607; PMID: 17652424
- Monier A, Claverie JM, Ogata H. Horizontal gene transfer and nucleotide compositional anomaly in large DNA viruses. BMC Genomics 2007; 8:456; http://dx.doi.org/10.1186/1471-2164-8-456; PMID: 18070355