
Abstract
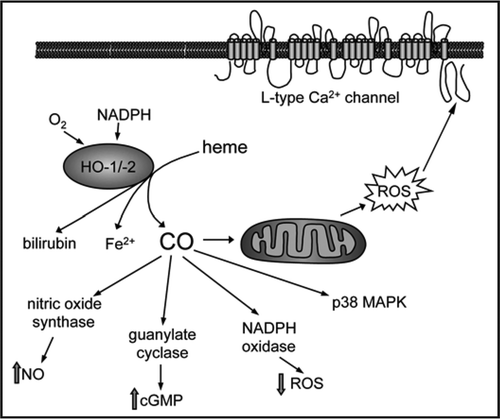
A wealth of recent studies has highlighted the diverse and important influences of carbon monoxide (CO) on cellular signaling pathways. Such studies have implicated CO, and the enzymes from which it is derived (heme oxygenases) as potential therapeutic targets, particularly (although not exclusively) in inflammation, immunity and cardiovascular disease.1 In a recent study,2 we demonstrated that CO inhibited cardiac L-type Ca2+ channels. This effect arose due to the ability of CO to bind to mitochondria (presumably at complex IV of the electron transport chain) and so cause electron leak which resulted in increased production of reactive oxygen species. These modulated the channel’s activity through interactions with three cysteine residues in the cytosolic C-terminus of the channel’s major, pore-forming subunit. Our study provided a potential mechanism for the cardioprotective effects of CO and also highlighted ion channels as a major potential target group for this gasotransmitter.
Acknowledgements
This work was supported by the British Heart Foundation and Wellcome Trust.
Figures and Tables
Figure 1 Schematic illustrating the synthesis and effects of carbon monoxide (CO). CO is generated by the O2 and NADPH-dependent catabolism of heme by HO-1 and HO-2. CO can stimulate or inhibit a number of signalling pathways, as illustrated. Of particular relevance to our studies, CO can bind to complex IV of the mitochondrial electron transport chain, leading to electron leak from complex III. This permits increased production of reactive oxygen species (ROS) that in turn leads to modulation of L-type Ca2+ channel activity via interaction with three cysteine residues located in the cytoplasmic C-terminal of the channel's α subunit.
Addendum to: