
Abstract
It has become increasingly clear that glucocorticoid (GC) signaling not only comprises classic nuclear receptor binding—that is, glucocorticoid receptors (GRs) to their response element in the nucleus—but also involves rapid, non-genomic efforts to regulate signaling cascades and other cell functions in the cytoplasm as well as other cell organelles. In a recent study, we found that GRs form a complex with B-cell lymphoma 2 (Bcl-2), translocate to mitochondria in response to corticosterone (CORT), and modulate mitochondrial calcium and oxidation in an inverted U–shaped manner. It is also well-established that steroid and thyroid hormone receptors regulate mitochondrial function to protect cells against various challenges and modulate synaptic plasticity. Here, we explore how such work reveals a fundamental mechanism whereby GCs regulate mitochondrial functions, and provides a mechanistic basis for therapeutic methods to rescue mitochondrial dysfunction during chronic stress or related psychiatric and neurodegenerative disorders.
Acknowledgements
We acknowledge the support of the Intramural Research Program of the NIMH. Ioline Henter provided outstanding editorial assistance.
Figures and Tables
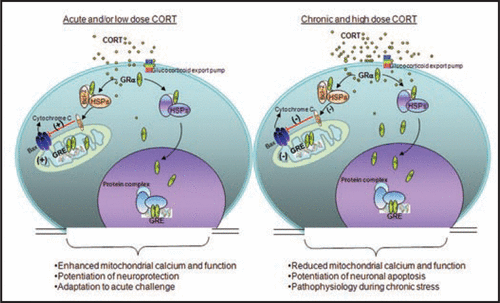
Figure 1 Biphasic effect of Glucocorticoids (GCs) in regulating mitochondrial function. GCs are secreted by adrenal glands in a circadian and stress-related fashion. GCs readily penetrate the cell membrane and interact with cytoplasmic glucocorticoid receptors (GRs). GRs travel to the nucleus to regulate gene expression by binding to glucocorticoid response element (GRE). Here, GRs formed a complex with the anti-apoptotic protein B-cell-lymphoma 2 (Bcl-2) in response to corticosterone (CORT) treatment, and translocated with Bcl-2 into mitochondria after acute treatment with low or high doses of CORT in primary cortical neurons; they also upregulated mitochondrial calcium levels, membrane potential and oxidation. However, after long-term (three days) treatment, high, but not low, CORT decreased GR and Bcl-2 levels in mitochondria. In addition, three independent measures of mitochondrial function—mitochondrial calcium holding capacity, mitochondrial oxidation and membrane potential—were also regulated by long-term CORT treatment in an inverted “U”-shape. Bcl-2 was able to inhibit the formation of Bax-containing pores on the mitochondrial outer membrane and reduced the release of calcium and cytochrome C from the mitochondria. This regulation of mitochondrial function by CORT correlated with neuroprotection; that is, treatment with low doses of CORT demonstrated a neuroprotective effect, whereas treatment with high doses of CORT enhanced kainic acid (KA)-induced toxicity of cortical neurons.
Addendum to: