Abstract
The Prion protein (PrP) is a membrane-tethered glycoprotein that plays a central role in a unique class of neurodegenerative diseases that affect humans and other mammals. Prion diseases have genetic and sporadic origins, but their infectious nature sets them apart from other neurodegenerative disorders. According to the ‘protein-only’ hypothesis, misfolded PrP conformers (prions) are responsible for both spongiform degeneration of the brain and disease transmissibility. Thus, understanding PrP conformational dynamics is key to developing effective therapies. Classic studies showing the different susceptibility to prion disease in mammals have recently found support in structural and transgenic studies with PrP from susceptible (mouse, hamster) and resistant (rabbit, horse, dog) animals. These studies identify key residues in PrP that determine both PrP structure and its propensity to acquire a ß-structure conformation proposed to be neurotoxic.
Prion diseases are transmissible neurodegenerative disorders characterized by spongiform vacuolation of brain neurons. It is now widely accepted that prion diseases are caused by the accumulation of misfolded conformers of the prion protein (PrP) in the brain.Citation1,Citation2 Native (cellular) PrPC is a soluble glycoprotein anchored to the membrane that possesses a flexible unstructured N-terminus and a globular C-terminus. The structure of the globular domain of human PrPC was resolved by X-ray crystallographyCitation3,Citation4 and nuclear magnetic resonance (NMR),Citation5 revealing three α-helices and two β-pleated domains. PrP causes several mammalian diseases because of its ability to undergo conformational changes, resulting in insoluble isoforms rich in β-pleated domains called “scrapie” PrP (PrPSc). PrPSc is resistant to denaturing agents and proteases, and is responsible for infectivity, whereas precursors or byproducts of PrPSc are proposed to be the neurotoxic species.Citation6 Although multiple conformational states have been resolved experimentally for PrP, cracking the code that governs PrP conversion has proved much tougher. In this regard, structure/function studies of natural variants have provided valuable information. Classic transmissibility experiments identified different susceptibility to prions: mice, rats and hamsters are easily infected with scrapie prions from sheep, but rabbits are resistant to human and sheep prions.Citation7,Citation8 Later studies indicated that intrinsic factors in rabbit PrP (its unique sequence, see ) dictate its inability to form pathogenic and/or infectious conformers.Citation9–Citation11 Dogs and horses also seem resistant to prions, although the experimental evidence in these cases is weaker because they are not acceptable experimental models. Thus, understanding how differences in PrP sequence translate into disease susceptibility could provide clues about the structural determinants in PrP. Several independent studies focusing on rabbit PrP have recently shed light into the molecular principles regulating PrP conversion and neurotoxicity in vitro and in vivo.
PrP Sequence and the β-state
To understand the structural peculiarities of the globular domain of PrP, Chakrabartty et al.Citation12 subjected recombinant PrP 90–231 from susceptible (Syrian hamster, mouse) and resistant (rabbit, horse and dog) mammals to comparative biochemical and structural studies (). Using hamster PrP, the authors first characterized a non-native, β-sheet rich conformer denoted β-state that appeared under moderately acidic pH and denaturing conditions (pH 4, 4 M urea). The β-state was distinguished from the native and unfolded states by applying circular dichroism (CD), a spectroscopic technique that associates differential absorption of circular polarized light with the secondary structure of proteins. Differential sedimentation indicated that the β-state was composed of both PrP monomers and octamers in equilibrium. Then, using an ingenious two-wavelength method of CD, the authors measured the relative amount of β-state for hamster, mouse, rabbit, horse and dog PrP under moderate to strong denaturing conditions (pH 7 to 4, urea 0 to 9 M). At neutral pH (7.0), none of the proteins populated the β-state independently of urea, but increasing acidic pH affected the five proteins differently. At pH 5.0, only hamster PrP populated the β-state, suggesting that this is the most unstable PrP structure. At pH 4.5, both hamster and mouse PrP accumulated large amounts of β-state, whereas rabbit PrP showed a small fraction of β-state. Under the most acidic conditions (pH 4.0), all of them populated the β-state, indicating that, when forced by the environment, all five proteins can misfold. However, horse and dog PrP had smaller fractions of β-state (60% and 10%, respectively) than hamster, mouse and rabbit (100%), demonstrating different propensities to populate the β-state. Interestingly, the propensity to form the β-state seemed to correlate with the disease susceptibility of each animal (hamster > mouse > rabbit > horse > dog), supporting old observations with new quantitative and comparative data.
To illustrate the importance of β-state PrP in prion diseases, the authors demonstrated its toxicity in cell culture. Rat Pheochromocytoma cells (PC12) treated with native mouse PrP exhibited normal viability. However, cells were efficiently killed with low µM concentrations of β-state mouse PrP,Citation12 indicating that the β-state is similar to β-structured oligomers of proteins involved in other neurodegenerative conditions, such as the Amyloid-α peptide (Alzheimer's disease) and α-Synuclein (Parkinson's disease).
PrP Sequence and in vivo Neurotoxicity
These detailed in vitro comparative studies provided experimental support for decades-old observations on disease susceptibility. However, in vivo evidence for the differential behavior of these proteins was still missing. In our own independent studies, we addressed that specific question by creating transgenic flies expressing wild type, full-length hamster, mouse and rabbit PrP.Citation13 Then, we compared the consequence of expressing each protein in biochemical, histological and behavioral assays. We had previously shown that hamster PrP progressively misfolds into PrPSc-like conformers that induce strong vacuolar changes in brain neurons.Citation14 In contrast, neither mouse nor rabbit PrP induced neurodegeneration in the brains of transgenic flies. This was the expected result for rabbit PrP, but it was a surprising result for mouse PrP, a susceptible species and a widely used model for PrP conversion. As a more direct test for neuronal dysfunction, we assayed the ability of the three proteins to induce progressive locomotor dysfunction. Both hamster and mouse PrP induced locomotor defects in a few days, whereas rabbit PrP was similar to control flies. Strikingly, mouse PrP was similar to hamster PrP in the locomotor assay, but similar to rabbit PrP in the progressive brain degeneration. We reasoned that the distinct neurotoxicity of hamster, mouse and rabbit PrP in flies should correlate with unique misfolding patterns of each protein. Although all three proteins accumulated in the detergent-insoluble fraction in older flies, only hamster and mouse PrP were recognized by a conformational antibody specific for PrPSc-like structures.Citation15 This inability to acquire a conformation relevant to PrP pathobiology clearly separated rabbit PrP from hamster and mouse PrP. To further distinguish the structural/biochemical properties of hamster and mouse PrP, we performed size fractionation of the misfolded species. Compared to mouse PrP, hamster PrP exhibited a higher propensity to form high molecular weight particles. Thus, our studies uncovered a strong correlation between the neurotoxic potential of hamster, mouse and rabbit PrP and their ability to misfold into disease-specific conformations (early neuronal dysfunction) and form large aggregates (late vacuolar degeneration).Citation13
Our observations in transgenic flies with full-length PrP are in strong agreement with the experiments of Chakrabartty et al. discussed above. Based on their description of octameric β-state as the PrP species responsible for cytotoxicity, we could venture to argue that the differential population of β-state accounts for the unique patterns of PrP aggregation and neurotoxicity in flies. We have observed hamster and mouse PrP oligomers in young flies by size exclusion chromatography, making β-state the ideal candidate for the early, aggressive locomotor dysfunction. Further aggregation of β-state oligomers might correspond to the high molecular weight particles observed in hamster PrP, making this higher order structures putatively responsible for the progressive spongiform degeneration. Thus, in vitro structural studies with recombinant, unglycosylated PrP90-231 yielded similar results to in vivo studies with full-length, glycosylated PrP, demonstrating the relevance and complementarity of multiple experimental platforms in addressing complex biological and biomedical questions.
The Global Reach of the Rigid Loop
A third paper has also focused on understanding the structural stability of rabbit PrP. Lin et al.Citation16 resolved the solution structure of rabbit PrP91-228 using NMR and compared it to the known PrP structures.Citation5,Citation17,Citation18 The most obvious difference in rabbit PrP is the presence of a continuous area of positive charges on both surfaces of PrP. Because intermolecular interactions are sensitive to surface charge, the unusual positive charges in rabbit PrP may inhibit cross-species PrP/PrP interactions or interaction with cofactors critical for prion conversion (protein-X or conversion factor).Citation19,Citation20 Additionally, this study produced detailed data on the dynamics of rabbit PrP that point to the highly organized state of the β2-α2 loop: (1) The backbone relaxation rate of the β2-α2 loop is similar to that of the ordered α1 and α2 domains, and unlike the other loops, which are unordered and more flexible; and (2) the amplitude of internal motions by residue (which measures local stability) in the β2-α2 loop is also similar to the structured regions. Thus, rabbit PrP possesses a highly ordered β2-α2 loop that may contribute to the local as well as global stability of the protein. To test this idea, the authors introduced the S174N replacement in the β2-α2 loop of rabbit PrP from mouse PrP (positions according to hamster PrP, see ). As expected, rabbit PrP[S174N] had profound effects on protein structure: (1) altered the charge distribution, both locally and globally, (2) reduced the number of hydrogen bonds, especially those involved in stabilizing the tertiary structure and (3) increased the local mobility of residues in the β2-α2 loop, but also affected the distant α3. These NMR studies emphasized the contribution of a highly ordered β2-α2 loop in dictating the global charge distribution and structural stability of rabbit PrP.
Office Solutions: Stapling Domains Together
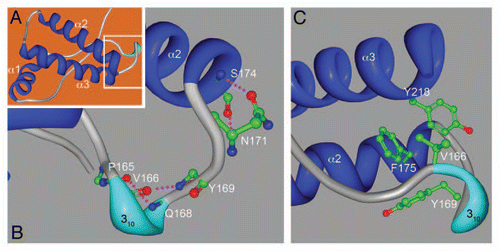
These three recent studies provide new experimental support for the unique structural and biological properties of rabbit PrP. But the precise mechanism that stabilizes rabbit PrP was only revealed thanks to X-ray crystallography. Chakrabartty et al. continued their conformational studies by resolving the X-ray crystal structure of natively folded rabbit PrP121-230.Citation12 This is no small feat given that the only X-ray crystal structures available to date are those of sheep and human PrP.Citation3,Citation4 The overall structure of rabbit PrP is very similar to human PrP (), as expected from NMR studies on several PrP species.Citation5,Citation17,Citation18 Crystallography confirmed the formation of a short 310 helix (also predicted by NMR) in the β2-α2 loop through backbone hydrogen bond exchanges between P165-D167 and V166-Q168 (). Closer to helix 2, though, the authors found a new structure formed by the double exchange of hydrogen bonds between the side chain of N171 and its backbone carbonyl with the backbone amide of S174 and its side chain hydroxyl (). In addition to these strong interactions, the flanking hydrophobic residues V166, Y169, F175 and Y218 form a hydrophobic pocket that stabilizes the β2-α2 loop, α2 and α3 domains (). Together, hydrophilic and hydrophobic interactions form a helix-capping motif with the evocative name of “hydrophobic staple” (). The S174 that provides a key hydroxyl for the second hydrogen bond is very rare in mammals (), suggesting that the newly reported “molecular staple” is directly responsible for the conformational stability of rabbit PrP. To prove the relevance of S174, the authors showed that the S174N mutation (the same as in the NMR studyCitation16) allowed rabbit PrP to populate the β-state under conditions of pH and urea in which wild type rabbit PrP exhibited native folding. Thus, X-ray crystallography identified a unique feature in rabbit PrP than explains its unusual biological properties.
Beyond the Rigid Loop
Is the presence of this molecular staple in the rigid loop enough to explain PrP neurotoxicity and disease susceptibility in other animals? Unfortunately, the residue that allows the formation of the molecular staple in the rabbit (S174) is not conserved in the horse and the dog (N174), the other prion-resistant animals. What other determinants of PrP structure are present in the horse and the dog? Horse PrP, like rabbit, possesses a well-defined β2-α2 by NMR,Citation21 but the loop sequences are very divergent. Horses and other equines contain two unique replacements in the loop (S167-E168, ), although S167 seems to be the most critical because of its ability to stabilize mouse PrP.Citation21 S167 seems to be in close contact with F225 (another exclusive substitution in the horse) in α3, providing a unique mechanism for stabilizing the β2-α2 loop and α3. Additionally, the Y225F substitution (Y225V in rabbit) introduces a hydrophobic residue that has to be buried in a hydrophobic pocket, which can also contribute to global stability, arguing for the β2-α2 loop/α3 interaction as key for PrP stability. In contrast to the horse, the sequence of the rigid loop in the dog is identical to mouse PrP, indicating that this region does not contribute to the conformational stability of dog PrP. Intriguingly, dog PrP possesses a few unique residues, including the acidic substitution D159 (E159 in greyhounds) in the previous loop (α1-β2). This loop is highly conserved among mammals, and only canines carry this substitution, suggesting that D159 may play a role in providing global stability to dog PrP. The NMR structure determined that dog PrP has an unusual charge distribution both locally and globally,Citation22 but no further details are available to explain the extreme stability of dog PrP. To help in this area, we are currently carrying out studies to determine the stabilizing effect of the N159D substitution in mouse PrP (manuscript in preparation).
Concluding Remarks
Continuing progress in understanding how the primary sequence dictates PrP structure will contribute to explaining PrP neurotoxicity and disease susceptibility. However, more progress is needed to gain a global understanding of PrP structure: how local changes affect distal domains. Whether the structural data comes from X-ray crystallography or NMR, experimental structure/function studies will continue to provide valuable knowledge about the rules governing PrP conversion. This step may be critical towards designing novel therapeutic approaches that block or revert PrP conversion and disease progression.
Figures and Tables
Figure 1 Sequence divergence in the globular domain. Alignment of the sequence of globular domain of PrP from Syrian hamster (accession number B34759), mouse (AAA39996), rabbit (AAD01554), horse (ACO71291) and dog (ACG59277). Amino acid numbering (bottom row) is indicated according to hamster PrP. The predicted secondary structures are shown on top: β-sheet (blue), α-helices (red/yellow), a short 310-helix (purple) and the unstructured loops are shown in gray. Alignment was performed using ClustalW (ebi.ac.uk/Tools/msa/clustalw2). Color-coding of amino acids: Red, small and/or hydrophobic; blue, acidic; magenta, basic; green, hydrophilic, charged. The double staple indicates the unique stabilizing mechanism of rabbit PrP.
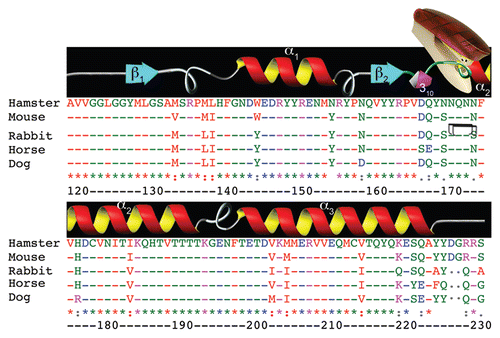
Figure 2 The “hydrophobic staple” in rabbit PrP. (A) 3-dimension structure of rabbit PrP as predicted by X-ray crystallography. The three α helices (α1–3, blue) are indicated. The area inside the white box is shown in more detail in (B and C). (B) Local hydrophilic interactions stabilize the β2-α2 loop and α2. Hydrogen bonds between backbone carbonyls of P165 and V166 and the amides of Q168 and Y169 to form a 310-helix (cyan) that forms a key turn in the loop. A double hydrogen bond between N171 and S174 (unique to rabbit) forms the helix-capping motif. (C) A hydrophobic pocket brings together the loop, α2 and α3, strengthening the helix-capping motif. The figure was created with Protein Workshop 3.9,Citation23 in the Protein Data Bank (pdb.org/pdb/home/home.do) from PDB ID: 3079.Citation12
Acknowledgements
This work was funded by the NIH grant R01NS062787, McGregor Foundation and the President's Discretionary Fund and Alliance BioSecure to W.Z., and the NIH grant DP2 OD002721-01 to P.F.F.
References
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 477
- Haire LF, Whyte SM, Vasisht N, Gill AC, Verma C, Dodson EJ, et al. The crystal structure of the globular domain of sheep prion protein. J Mol Biol 2004; 336:1175 - 1183
- Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol 2001; 8:770 - 774
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 2000; 97:145 - 150
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930 - 936
- Barlow RM, Rennie JC. The fate of ME7 scrapie infection in rats, guinea-pigs and rabbits. Res Vet Sci 1976; 21:110 - 111
- Gibbs CJ Jr, Gajdusek DC. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science 1973; 182:67 - 68
- Vorberg I, Groschup MH, Pfaff E, Priola SA. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J Virol 2003; 77:2003 - 2009
- Courageot MP, Daude N, Nonno R, Paquet S, Di Bari MA, Le Dur A, et al. A cell line infectible by prion strains from different species. J Gen Virol 2008; 89:341 - 347
- Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, Lehmann S, et al. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci USA 2001; 98:4055 - 4059
- Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, et al. Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci USA 2010; 107:19808 - 19813
- Fernandez-Funez P, Zhang Y, Casas-Tinto S, Xiao X, Zou WQ, Rincon-Limas DE. Sequence-dependent prion protein misfolding and neurotoxicity. J Biol Chem 2010; 285:36897 - 36908
- Fernandez-Funez P, Casas-Tinto S, Zhang Y, Gomez-Velazquez M, Morales-Garza MA, Cepeda-Nieto AC, et al. In vivo generation of neurotoxic prion protein: role for hsp70 in accumulation of misfolded isoforms. PLoS Genet 2009; 5:1000507
- Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 1997; 390:74 - 77
- Wen Y, Li J, Yao W, Xiong M, Hong J, Peng Y, et al. Unique structural characteristics of the rabbit prion protein. J Biol Chem 2010; 285:31682 - 31693
- Lopez Garcia F, Zahn R, Riek R, Wuthrich K. NMR structure of the bovine prion protein. Proc Natl Acad Sci USA 2000; 97:8334 - 8339
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121–321). Nature 1996; 382:180 - 182
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83:79 - 90
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA 1997; 94:10069 - 10074
- Perez DR, Damberger FF, Wuthrich K. Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol 2010; 400:121 - 128
- Lysek DA, Schorn C, Nivon LG, Esteve-Moya V, Christen B, Calzolai L, et al. Prion protein NMR structures of cats, dogs, pigs and sheep. Proc Natl Acad Sci USA 2005; 102:640 - 645
- Moreland JL, Gramada A, Buzko OV, Zhang Q, Bourne PE. The Molecular Biology Toolkit (MBT): a modular platform for developing molecular visualization applications. BMC Bioinformatics 2005; 6:21