Abstract
Presenilin (PS) mutations are the main cause of Familial Alzheimer’s Disease (FAD), and have been demonstrated to cause an imbalance of intracellular Ca2+ homeostasis. Though PS1 and 2 are generally considered to behave similarly in terms of their effects on Ca2+ handling, we have recently described a novel function, which is unique to PS2, i.e. the modulation of ER-mitochondria juxtaposition. Accordingly, PS2, but not PS1, affects the Ca2+ cross-talk between these organelles, a key feature in determining cell fate. In particular, PS2 over-expression, and more drastically that of FAD-linked PS2 mutants, strongly increases the interaction between ER and mitochondria, thus facilitating mitochondrial Ca2+ uptake. The likely mechanisms behind this phenomenon and its potential effects in cell physiology and pathology are discussed.
Presenilins 1 and 2 (PS1, PS2) are the catalytic core of γ-secretase, the enzymatic complex responsible for the final step in amyloid-beta (Aβ) peptides' production and mutations in their genes are the main cause of Familial Alzheimer's Disease (FAD).Citation1,Citation2
Presenilin is a nine trans-membrane domain protein that, once incorporated into the γ-secretase complex, rapidly undergoes endoproteolysis at its large cytosolic loop, generating two fragments (N- and C-terminal) which are considered the mature form of PS. Both PSs are mainly found at the level of Endoplasmic Reticulum (ER) membranes.Citation1–Citation3 Of interest, recent findings indicate that PSs are enriched in ER membrane patches closely associated to mitochondria,Citation4 the so called “Mitochondria-Associated Membranes (MAMs).”Citation5
Apart from their function in Aβ pep-tides' production, PSs appear involved in several physiological processes within the cell, including Ca2+ homeostasis, protein transport and turnover, autophagy, cell adhesion, neurotransmitter release and axon guidance.Citation6–Citation11
The highly homologue PS1 and PS2 are commonly thought to have almost overlapping cellular functions, with minor differences, e.g., PS1 is considered the more abundant and the main protein involved in Aβ peptides' generation.Citation2,Citation12 However, we have recently described a novel cell function of PS2 not shared by PS1, i.e., the modulation of ER and mitochondria interactions and thus, of their Ca2+ crosstalk.Citation13
ER and mitochondria are two major players in cellular Ca2+ homeostasis and their interaction is critical in cell physiology. Mitochondria are, in fact, known to massively take up Ca2+ when cytosolic microdomains of high Ca2+ concentrations (Ca2+ hot spots) are generated at the level of their outer membrane (OMM).Citation14 This type of events occur upon opening of either plasma membrane voltage-gated Ca2+-channels or ER Ca2+ releasing channels.Citation15,Citation16 Mitochondrial functions are in turn tightly regulated by the intramitochondrial Ca2+ concentration that is known to modulate few key dehydrogenases and, when the increases are too large or prolonged, to trigger apoptosis.Citation17,Citation18 Thus, Ca2+ cross-talk between ER and mitochondria contributes to cell fate determination and alterations in the two organelles' axis can dramatically affect cell state.Citation19
The number of proteins supposed to influence ER-mitochondria interactions is progressively increasing, among them the best studied include PACS-2,Citation20 Mfn-2,Citation21 IP3R-grp75-VDACCitation22 and Fis1-Bap31Citation23 complexes, and Trichoplein/mitostatin.Citation24 PS2 appears now as a novel member of this group:Citation13 in fact, by PS2 overexpression or downregulation, we have demonstrated that this protein (but not its homologue PS1) modulates the ER-mitochondria tethering by increasing the number and/or the extent of their contact sites. In particular, PS2 overexpression facilitates mitochondrial Ca2+ uptake following ER Ca2+ release, while the opposite occurs upon its downregulation. PS2 overexpression is thus associated to a higher probability of cytosolic Ca2+ hot spots generation at the level of the OMM. Strikingly, expression of FAD-linked PS2 mutations leads to a larger increase in ER-mitochondria interactions, compared to the wt counterpart, as demonstrated both in cell lines and in primary cortical neurons, a finding that may be of major importance for interpreting the role of Ca2+ dysregulation in AD ().
As far as the molecular mechanism behind this PS2 effect is concerned, available data are still insufficient to draw a clear conclusion and only hypotheses can be made:
(1) Mfn-2,Citation21 the protein that directly bridges, by homotypic interaction, ER and mitochondria, seems not to be directly involved since PS2 overexpression or downregulation does not modulate its protein level.Citation13
(2) PS2 could be required for the proper docking of a protein—one of its substrates—that in turn acts as a tether between the two organelles. It could be speculated that under normal conditions, wt PS2, as the catalytic core of the γ-secretase complex, cleaves most of this substrate, while FAD-PS2 mutants, which are known to have a reduced enzymatic activity,Citation25 leave un-cleaved a larger fraction of this tether, thus increasing the vicinity of the two organelles. However, the finding that pharmacological γ-secretase inhibition does not mimic this PS2 effectCitation13 argues against a major role of substrate cleavage in the increased interaction between ER and mitochondria.
(3) Since mitochondrial dynamics within the cell is regulated by Ca2+,Citation26 the increased ER-mitochondria interaction could be also due to an alteration of mitochondria movements due to the imbalance of ER Ca2+ handling caused by FAD-PS2 expression (reviewed in refs. Citation6, Citation7, Citation27 and Citation28). However, different treatments that alter ER Ca2+ concentration ([Ca2+]ER) do not mimic the PS2 effect on organelles' juxtaposition.Citation13
(4) The full length form of PS2 is likely involved in this phenomenon since a PS2 mutant devoid of endo-proteolytic maturation (PS2-D366A) is fully capable of increasing ER-mitochondria interactions.Citation13
(5) Based on quantitative considerations,Citation13 PS2 likely requires a partner protein to exert its effect in bridging ER and mitochondria; the nature of this partner is currently not clear, as it could be a molecule residing on ER and/or mitochondria membranes as well as a cytosolic component.
(6) Albeit highly homologous, the two PSs do not share this function and interestingly, the region with the highest variability between PS1 and PS2 is the big cytosolic loop, which is disrupted upon maturation;Citation1 this suggests that the PS2 cytosolic loop could be the region of the protein involved in the interaction with the hypothetical molecular partner and thus required for modulating ER-mitochondria juxtaposition.
Concerning the importance and significance of this effect in cell physiology and pathology, several considerations are allowed (). PS2 decreases [Ca2+]ER more potently than PS1,Citation27,Citation28 yet an additional effect on intracellular Ca2+ handling, i.e., the facilitation of ER-mitochondria Ca2+ transfer, was described only for PS2.Citation13 A decrease in [Ca2+]ER is generally considered protective against apoptosis since reduced [Ca2+]ER would decrease the probability of mitochondria Ca2+ overload;Citation17,Citation19,Citation29 on this line, the lower [Ca2+]ER due to expression of FAD-linked PS2 mutants, compared to those of PS1, could explain the milder AD phenotype described in patients bearing PS2 rather than PS1 mutations, although both groups show amyloid pathology.Citation1,Citation2 On the other side, reduced [Ca2+]ER could impair the physiological Ca2+ cross-talk between ER and mitochondria,Citation30 that is required for the proper activity of the latter organelle.
This novel effect of PS2 on ER-mitochondria juxtaposition adds further complexity to the scenario of Ca2+ dysfunctions due to FAD-linked PS mutations: on one hand, the increased ER-mitochondria interactions caused by PS2 mutants could represent a compensatory phenomenon, ensuring proper Ca2+ signals towards mitochondria, despite the decrease in [Ca2+]ER (); on the other hand, this effect could overcome the reduction in [Ca2+]ER and increase the probability of toxic Ca2+ transfer to mitochondria, with detrimental consequences on cell viability (). Again, the observed milder AD phenotypes linked to FAD-PS2 mutations could argue against this second possibility that considers PS2 mutations as more noxious than those in PS1. Interestingly, a much larger number of mutations in PS1 than in PS2 have been so far identified in FAD patients;Citation1 an odd difference for two such homologous proteins, suggesting that some PS2 mutations might be incompatible with life.
These different hypotheses can be now tested experimentally but nonetheless they highlight the complexity of Ca2+ dysfunction in neurodegeneration and the multifaceted nature of PSs.
Figures and Tables
Figure 1 ER-mitochondria juxtaposition and high Ca2+ microdomains on the cytosolic surface of OMM in SH-SY5Y cells. Cells were transfected with cDNAs coding for a FAD-PS2 mutant (PS2-T122R, B) or void vector (Control, A), together with those coding for a red mitochondrial and a green ER markers (mit-RFP and ER-GFP, respectively; upper cell in each part), in order to visualize ER-mitochondria juxtaposition sites (yellow pixels); increased number of interactions is observed in FAD-PS2 expressing cells. Alternatively, cells were transfected with cDNAs coding for a FAD-PS2 mutant (PS2-T122R, B) or void vector (Control, A) and the cameleon Ca2+ probe N33-D1cpv, targeted to the OMM,Citation15 and then analyzed to detect the generation of high Ca2+ microdomains on the cytosolic surface of OMM (yellow-to-red spikes) upon ER Ca2+ release induced by bradykinin (lower cell in each part). At a similar average cytosolic Ca2+ rise, higher and more abundant Ca2+ microdomains are generated close to the OMM in the FAD-PS2 expressing cell compared to the control one. See reference Citation13 for details.
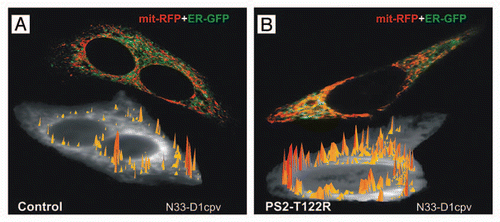
Figure 2 Model depicting the possible effects of FAD-PS2 mutations on ER-mitochondria interactions and Ca2+ cross-talk. In physiological condition (A), mitochondria receive constant Ca2+ signals from ER that ensure their proper activity.Citation30 Expression of FAD-PS2 mutants (B and C) decreases [Ca2+]ERCitation27,Citation28 and recruits mitochondria closer to ER Ca2+ releasing sites.Citation13 As a consequence, closer ER-mitochondria juxtaposition may compensate [Ca2+]ER reduction allowing mitochondria to receive appropriate Ca2+ signals that drive AT P production (B); alternatively, closer ER-mitochondria juxtaposition may expose mitochondria to excessive Ca2+ stimulation, triggering the apoptotic cascadeCitation29 by mitochondria Ca2+ overload, outer membrane permeabilization and release of pro-apoptotic factors (C).
![Figure 2 Model depicting the possible effects of FAD-PS2 mutations on ER-mitochondria interactions and Ca2+ cross-talk. In physiological condition (A), mitochondria receive constant Ca2+ signals from ER that ensure their proper activity.Citation30 Expression of FAD-PS2 mutants (B and C) decreases [Ca2+]ERCitation27,Citation28 and recruits mitochondria closer to ER Ca2+ releasing sites.Citation13 As a consequence, closer ER-mitochondria juxtaposition may compensate [Ca2+]ER reduction allowing mitochondria to receive appropriate Ca2+ signals that drive AT P production (B); alternatively, closer ER-mitochondria juxtaposition may expose mitochondria to excessive Ca2+ stimulation, triggering the apoptotic cascadeCitation29 by mitochondria Ca2+ overload, outer membrane permeabilization and release of pro-apoptotic factors (C).](/cms/asset/976cce8d-e08c-445e-97b2-896765ab419f/kcib_a_10915160_f0002.gif)
Addendum to:
References
- De Strooper B, Annaert W. Novel research horizons for presenilins and γ-secretases in cell biology and disease. Annu Rev Cell Dev Biol 2010; 26:235 - 260
- Vetrivel KS, Zhang YW, Xu H, Thinakaran G. Pathological and physiological functions of presenilins. Mol Neurodegener 2006; 1:4
- Laudon H, Hansson EM, Melén K, Bergman A, Farmery MR, Winblad B, et al. A nine-transmembrane domain topology for presenilin 1. J Biol Chem 2005; 280:35352 - 35360
- Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 2009; 175:1810 - 1816
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol 2009; 19:81 - 88
- Bojarski L, Herms J, Kuznicki J. Calcium dysregulation in Alzheimer's disease. Neurochem Int 2008; 52:621 - 633
- Mattson MP. ER calcium and Alzheimer's disease: in a state of flux. Sci Signal 2010; 3:10
- Parks AL, Curtis D. Presenilin diversifies its portfolio. Trends Genet 2007; 23:140 - 150
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146 - 1158
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, et al. Presenilins are essential for regulating neurotransmitter release. Nature 2009; 460:632 - 636
- Bai G, Chivatakarn O, Bonanomi D, Lettieri K, Franco L, Xia C, et al. Presenilin-dependent receptor processing is required for axon guidance. Cell 2011; 144:106 - 118
- Lai MT, Chen E, Crouthamel MC, DiMuzio-Mower J, Xu M, Huang Q, et al. Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J Biol Chem 2003; 278:22475 - 22481
- Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci USA 2011; 108:2777 - 2782
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998; 280:1763 - 1766
- Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, et al. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell 2010; 38:280 - 290
- Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 2010; 39:121 - 132
- Pizzo P, Pozzan T. Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol 2007; 17:511 - 517
- Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: the calcium connection. Biochim Biophys Acta 2010; 1797:607 - 618
- Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 2006; 174:915 - 921
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 2005; 24:717 - 729
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008; 456:605 - 610
- Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 2006; 175:901 - 911
- Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 2011; 30:556 - 568
- Cerqua C, Anesti V, Pyakurel A, Liu D, Naon D, Wiche G, et al. Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep 2010; 11:854 - 860
- Shen J, Kelleher RJ III. The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA 2007; 104:403 - 409
- Liu X, Hajnóczky G. Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int J Biochem Cell Biol 2009; 41:1972 - 1976
- Brunello L, Zampese E, Florean C, Pozzan T, Pizzo P, Fasolato C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J Cell Mol Med 13:3358 - 3369
- Zatti G, Burgo A, Giacomello M, Barbiero L, Ghidoni R, Sinigaglia G, et al. Presenilin mutations linked to familial Alzheimer's disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006; 39:539 - 550
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008; 27:6407 - 6418
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270 - 283