Abstract
Molecular geneticists tend to conceptualize disease pathogenesis from the mutated gene outward, an approach that does not take into account the impact of barrier requirements in determining disease phenotype. An ‘outside-to-inside’ perspective, which takes full advantage of the same genetic information, has provided quite different explanations for the ichthyoses, including several of the disorders of distal cholesterol metabolism. Elucidation of responsible pathogenic mechanisms also is pointing to appropriate, pathogenesis (pathway)-based therapeutic strategies. In the case of the lipid metabolic disorders, it takes full advantage of new molecular, genetic and cellular pathogenesis information to correct or bypass the metabolic abnormality. This approach fully exploits the unique accessibility of the skin to a topical approach. Moreover, since it will utilize topical lipids and lipid-soluble, and often generic, lipid-soluble drugs, these treatments should be readily transported across the stratum corneum. If successful, this approach could initiate an entirely new departure for the therapy of the ichthyoses. Finally, because these agents are relatively safe and inexpensive, this form of treatment has the potential to be widely-deployed, even in the developing world.
Introduction
Current treatment of the ichthyoses remains symptomatic, and largely directed towards reducing the scaling component of these disorders. Yet, such therapy of the ichthyoses is often irrational, because removal of excess scale can interfere with homeostatic responses that allow patients to survive in a harsh, terrestrial environment. Moreover, the favored alternative, corrective gene therapy, though seductive in concept, remains a distant dream, impeded by: (1) difficulties in transcutaneous drug delivery; (2) enormous costs of the required ‘designer gene’ approach; (3) discomfort of intracutaneous injections; and (4) unknown, long-term risks of transfection with viral vectors.
All ichthyoses, including inherited syndromic disorders of distal cholesterol metabolism, display a permeability barrier abnormality, with the severity of the clinical phenotype paralleling the prominence of the barrier abnormality (reviewed in ref. Citation1–Citation3). In our research, we have assumed that the cutaneous phenotype represents a best attempt by a metabolically-compromised epidermis to generate a competent permeability barrier in the desiccating, terrestrial environment (op. cit.). While “normal” epidermis mounts a vigorous, metabolic response in response to a compromised barrier that rapidly restores function,Citation4–Citation6 “ichthyotic” epidermis only partially succeeds in normalizing function.Citation1–Citation3 As a result, the clinical phenotype reflects the negative consequences of the genetic mutation for epidermal function, coupled with the epidermis' impaired, homeostatic response. Thus, unraveling the cellular and biochemical mechanisms that account for the barrier abnormality provides an explanation for the pathogenesis of the cutaneous phenotypes (reviewed in ref. Citation2, Citation3 and Citation7), and it could point to potentially-novel, pathway-based therapies. In disorders due to impaired cholesterol synthesis, evidence to date suggests that the clinical phenotype in most cases reflects either accumulation of toxic metabolites and/or deficiency of pathway end-product.Citation8 Moreover, in all of the lipid-metabolic disorders, whether due to metabolite accumulation, pathway product deficiency, or both, lamellar/non-lamellar (L/NL) phase separation within the lamellar bilayers accounts, at least in part, for the barrier abnormality (examples of pathogenic mechanisms for disorders of distal cholesterol metabolism are shown in ).
We typically assess four different, functional end-points; i.e., TEWL, pH, hydration and stratum corneum (SC) integrity, which separately or together can impact permeability barrier function. For example, a less cohesive SC, which often is due to an elevated pH of SC, increases proteolytic degradation of corneodesmosomes, which results in a poor quality SC. A high pH also activates serine proteases, which degrade lipid processing enzymes. Finally, the pH-driven increase also activates pro-inflammatory cytokines, such as IL-1α and IL-1β, further aggravating barrier function while provoking inflammation. By measuring multiple functional, structural and biochemical parameters and their structural/biochemical basis, we have been able to assemble a pathogenic composite for each disease. Together, this approach has identified key pathophysiologic abnormalities (e.g., metabolite accumulation and/or product depletion) in these disorders, which in turn could point to the most-promising, potential, pathogenic-based therapeutic interventions (see below).
Pathogenesis of Multisystem, Cholesterol Biosynthetic Disorders
Nine enzymatic steps are required to generate cholesterol from lanosterol, with the further generation of cholesterol sulfate from cholesterol comprising a tenth step (). While syndromic disorders, with a variety of developmental malformations, have been reported in seven of these diseases,Citation9–Citation13 an abnormal cutaneous phenotype has been described in only six, i.e., lathosterolosis, desmosterolosis, Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects (CHILD) syndrome, Conradi-Hünermann-Happle syndrome (CHH) or X-linked chondrodysplasia punctata type 2 (CDPX2), SC4MOL deficiencyCitation14–Citation16 and X-linked ichthyosis (XLI).Citation17 The pathogenesis of the ichthyosiform dermatosis (and likely the extracutaneous abnormalities) in all of the inborn errors of distal cholesterol metabolism can be variously attributed to either: deficiency of cholesterol in cell membranes and/or toxic effects of accumulated sterol precursors with resulting functional alterations.Citation18,Citation19 Sterol precursors can only partially substitute for cholesterol in the formation of SC lamellar membranes, and cholesterol is one of the three key SC lipids (along with ceramides and free fatty acids) required to form lamellar membranes (reviewed in ref. Citation20). Additional, downstream pathogenic mechanisms whereby sterol metabolites could contribute to disease pathogenesis include: (1) formation of oxysterol metabolites that either downregulate cholesterol synthesis or activate the liver X receptor;Citation21 (2) altered hedgehog pathway signaling (HOX normally is tethered onto cell membranes via a cholesterol moiety);Citation22,Citation23 and/or (3) deficient peroxisomal function, as we have described in both CHH, CHILD patients and in the ‘bare patches’ mouse model,Citation24–Citation28 which displays Nsdhl mutations that mimic CHILD syndromeCitation29,Citation30 (further information about our work on the pathogenesis of CHILD syndrome, CHH and XLI is provided below); and (4) sterol metabolite-accelerated degradation of HMGCoA reductase.Citation31
While ichthyosis is not clinically apparent in Smith-Lemli-Opitz syndrome (SLOS) (OMIM #270400) [7-dehydrocholesterol reductase (DHCR7) deficiency], both photosensitivity and a propensity to develop eczema are commonCitation32 (Drs. Rosalind Elias and R. Steiner, personal communication). DHCR7 deficiency impairs both desmosterol and 7-dehydrocholesterol metabolism,Citation20,Citation21,Citation33 resulting in elevated 7-DHC and 8-DHC blood levels, with proportionate, phenotype-dependent reductions in serum cholesterol,Citation9–Citation13,Citation24,Citation32,Citation34,Citation35 alterations that are mimicked in Dhcr7−/− and +/− mice,Citation36 and in mice with a knock-in of the human T93M mutation.Citation37 SLOS is fairly common (predicted incidence of ≈1:10,000),Citation8 with over 120 different DHCR7 mutations identified to date.Citation32
Although the PI is unaware of lathosterolosis cases in the US, several patients have been described in Europe, and all have prominent ichthyosis. Moreover, a mouse model of lathosterolosis (Sc5d−/− and +/−) is available,Citation15 which should allow assessment of pathogenic mechanisms in this disorder (see below). Furthermore, a US kindredCitation38 and several additional patients in EuropeCitation14 have been described with desmosterolosis, a disorder that displays prominent congenital anomalies, but minimal evidence of skin abnormalities. Finally, a prominent skin phenotype has been described in two patients with SC4MOL deficiency, who present with a severe ichthyosiform dermatosis and psoriasiform features.Citation16
Conradi-Hünermann-Happle Syndrome (CHH) or X-linked dominant chondrodysplasia punctata type 2 (CDPX2) (OMIM #302960) exhibits linear bands of scaling or follicular spikes in a morphogenic pattern (i.e., along the lines of Blaschko), and generalized erythroderma, most prominently in neonates. Involved skin sites conform to regions in which the mutant X-chromosome predominates.Citation39,Citation40 The cutaneous features of CHH and CHILD syndrome can improve after infancy,Citation40 due to diminished viability of keratinocytes bearing the mutant X chromosome.Citation29 The cutaneous phenotype in CHILD syndrome (OMIM #308050), however, differs in its distribution from CHH; i.e., it is strictly unilateral, including both skeletal defects and internal organ involvement. Skin lesions are circumscribed plaques, surmounted by prominent wax-like scales, which typically involve flectures.Citation23
CHH is caused by mutations in EBP (emopamil-binding protein) that encodes 3β-hydroxysterol-Δ8, Δ7-isomerase, which catalyzes the conversion of 8(9)-cholestenol to lathostero1,Citation34,Citation41,Citation42 resulting in diagnostic elevations in serum 8-dehydrocholesterol and 8(9) cholesterol.Citation43 Mutations in NSDHL, which encodes a member of the enzyme complex that removes the C-4 methyl group from lanosterol, underlie CHILD syndrome. However, CHILD syndrome can also be caused by mutations in EBPCitation9,Citation43 (). Given the close proximity of the sites of metabolic blockade, the presence of some phenotypic overlap between CHILD and CHH is not surprising (reviewed in ref. Citation9). Availability of mouse models of CHH and CHILD syndromeCitation8 should allow for an assessment of the mechanistic basis for the cutaneous phenotype in CHH and CHILD syndrome, and for preclinical evaluation of potential therapies for these patients.
Both the density of lamellar bodies (LB) and LB secretion appear normal in CHH, but organelle contents are abnormal, displaying vesicular inclusions. Moreover, newly-secreted material fails to disburse at the stratum granulosum-SC interface.Citation7 Furthermore, these electron-lucent vesicles persist as discrete spheres after secretion at the stratum granulosum-SC interface. Importantly, maturation of lamellar bilayers is delayed, and bilayer membranes with normal morphology are displaced by extensive areas of lamellar/non-lamellar phase separation ().Citation29 Yet, the morphology of clinically-affected skin sites in CHILD syndrome is even more dramatically abnormal than that in CHH. Although LB form normally, they display almost no internal lamellae, and they fuse into intracellular multivesicular bodies, which then are largely (but incompletely) secreted.Citation7 Nevertheless, the SC displays a huge expansion of the extracellular matrix, which is filled with interspersed lamellar and non-lamellar material.Citation7 Together, these features predict a severe barrier abnormality in both CHILD and CHH.
Recessive X-linked ichthyosis (XLI).
The pathogenesis of XLI is better known than for any of the other ichthyoses. As a result of steroid sulfatase (SSase) deficiency in XLI, cholesterol sulfate (CSO4) accumulates in the outer epidermis,Citation44–Citation46 in erythrocyte cell membranes,Citation45,Citation47 as well as in both the LDL (β-lipoprotein) and pre-LDL fractions of plasma.Citation45 But CSO4 levels in epidermis are an order of magnitude higher than are levels in blood,Citation45,Citation47 likely explaining the prominence of epidermal vs. other organ involvement in XLI.Citation48 Normally, CSO4 levels decline to about 1% of lipid mass in the outer SC, through ongoing hydrolysis during SC transit.Citation49,Citation50 In contrast, the SC in XLI typically contains 10–12% cholesterol sulfate (by dry weight).Citation48 Hydrolysis of CSO4 generates some of the cholesterol required for the barrier, while conversely, CSO4 itself is a potent inhibitor of HMGCoA reductase, further reducing cholesterol levels in XLI.Citation48 Although SSase is secreted from lamellar bodies (like other lipid hydrolases that process barrier lipid precursors into their own hydrophobic products), CSO4 is delivered to the SC interstices by its extreme amphiphilicity, which allows it to diffuse readily across cell membranes;Citation51 i.e., in the absence of a lipid milieu within corneocytes, cholesterol sulfate likely partitions preferentially into the highly hydrophobic, extracellular domains of the stratum corneum. Accumulation of CSO4, coupled with cholesterol depletion, provokes lamellar/non-lamellar phase separation,Citation17 accounting for the barrier abnormality in XLI.Citation52
Recent Studies in Relevant Animal Models
Insig-2 (Epi-insig) DKO mice.
The Brown and Goldstein group recently published relevant work on another animal model with aberrant cholesterol synthesis; i.e., epidermal-localized Insig-1 mice with an additional germ-line deletion of Insig-2 (Epi-Insig) DKO mice.Citation53 This model mimics the syndromic human disorder, Ichthyosis Follicularis, Atrichia and Photophobia (IFAP) syndrome (OMIM #308205). Deletion of these intracellular proteins allows migration of SREBP from the endoplasmic reticulum to the Golgi apparatus followed by translocation to the nucleus, where these SREBPs excessively stimulate several genes involved in cholesterol synthesis. As a result, both sterol metabolites and cholesterol accumulate in the skin. Pertinently, these mice respond to treatment with topical simvastatin, which simultaneously normalizes both metabolite production and cholesterol levels in epidermis.Citation53 This recent work, coupled with preliminary evidence of efficacy with topical lovastatin plus cholesterol in CHILD syndrome,Citation54 provides evidence that metabolite accumulation in the epidermis can be toxic; and conversely, that blockade of metabolite production (and normalization of cholesterol levels) could be beneficial for these disorders.
Mouse models of SLOS.
While SLOS patients display a minimal skin phenotype, Dhcr7−/− mice display prominent ichthyosis, with neonatal lethality due to a putative permeability barrier abnormality.Citation36 More pertinent to SLOS patients, who display residual enzyme function, our still-unpublished preliminary studies in Dhcr7+/− mice (with S. Patel) demonstrate epidermal structural abnormalities in both lamellar body contents and lamellar bilayer organization, predictive of a barrier abnormality in SLOS patients with comparable reductions in enzyme activity. These mice also display serum 7DHC and cholesterol levels that are comparable to patients with moderate SLOS due to partial loss-of-function mutations.Citation36 One relatively-common mutation in SLOS (T93M) has been recapitulated in a transgenic ‘knockin’ mouse model.Citation37,Citation55 While their skin phenotype has not yet been assessed, this model should also be useful to assess both pathogenic mechanisms and preclinical therapeutic studies.
Mouse models of CHH, CHILD, desmosterolosis and lathosterolosis.
We also have recently begun to assess a potential animal model of desmosterolosis. Like Dhcr7−/− mice, Dhcr24−/− mice are neonatal lethal, apparently due to a failure of epidermal development in utero, but possibly also due to a barrier defect.Citation56,Citation57 In contrast, Dhcr24+/− mice survive, and like Dhcr7+/− mice, they show structural evidence of a skin barrier abnormality. While the Bpa, Stri and Tattered strains closely mimic varying severities of CHH and CHILD syndromes, one of these animal models is not a pure analogue of human disease; i.e., the Sc5d−/− mouse, but partial loss-of-function in the Sc5d+/− could reflect comparable reductions in enzyme function in lathosterolosis. Thus, this model could also be useful to assess pathogenic mechanisms and potential therapies for lathosterolosis.
Therapeutic Implications
Current therapy of the ichthyoses is largely aimed at scale removal; i.e., it is purely symptomatic. Not only is this form of therapy relatively ineffective, it often can be counterproductive; e.g., by removing ‘excess’ stratum corneum (SC), symptomatic therapy can do more harm than good (=‘therapeutic paradox’) as seen with: (1) retinoids in the treatment of Netherton syndrome (NS),Citation58 and sometimes in epidermolytic ichthyosis (EI);Citation59 (2) excessive absorption of salicylates, lactic acid or immunosuppressive molecules in NS;Citation58 and (3) worsening of the permeability barrier after alpha-hydroxyacid applications, accentuating fluid and electrolyte abnormalities in HI, NS, EI and other disorders.Citation58 While localized gene therapy has shown promise in focal conditions, such as pachyonychia congenital,Citation60 generalized skin involvement in most of the ichthyoses makes topical gene therapy impractical. Moreover, this approach can be limited by pain from injections,Citation60 unknown risks for viral vector-induced neoplasia, and the high cost of ‘designer gene’ replacement, which together render gene therapy largely impractical for this diverse group of disorders, and certainly unattainable for patients in the developing world.
Very recently, we proposed a novel, pathogenesis-based approach to the treatment of ichthyosis in disorders of distal cholesterol metabolism;Citation54 i.e., provision of pathway product plus blockade of metabolite production, exploiting this new understanding of disease pathogenesis (). If metabolite accumulation appears to account for the cutaneous phenotype, it would seem reasonable to deploy a proximal enzyme inhibitor [e.g., of HMGCoA reductase (lovastatin)] to reduce levels of potentially-toxic metabolites. In addition, provision of the pathway product (cholesterol) to avoid epidermal dysfunction due to cholesterol deficiency could also be beneficial by further downregulating (negative feed-back) of HMGCoA reductase activity. Then, this new information about disease pathogenesis has the potential to be deployed rapidly into topical therapy for patients with rare inherited disorders of distal cholesterol metabolism (). Another not-so-rare, inherited disorder of distal cholesterol metabolism, X-linked ichthyosis (XLI), occurs in 1:2,000–6,000 males. Our studies over several years already have shown that disease pathogenesis in XLI also reflects both metabolite (CSO4) accumulation and end-product (cholesterol) deficiency (see also above).Citation17,Citation46,Citation48 Pertinently, topical cholesterol alone in our experience is not effective in XLI, but the ichthyosis in XLI could be treatable with either a topical statin or a sulfotransferase inhibitor (plus cholesterol). It must be emphasized that blockade of metabolite production alone, though it could be temporarily useful, cannot be utilized as monotherapy for the cutaneous phenotype in these disorders, because the end-product of this pathway (i.e., cholesterol) is required to prevent development of a permeability barrier abnormality (topical statins provoke a barrier abnormality with ichthyosiform changes in normal mouse skin).Citation61,Citation62
In very preliminary studies, we have treated two patients with one of the rare disorders of distal cholesterol metabolism (CHILD syndrome) [Drs. Amy Paller (Northwestern University) and Marina Rodriquez-Martin (Canary Islands Univ Hospital)—point mutation in G83Dp Gly83 Asp in NSHDL, and nonsense mutation (c.317C>A; p.S106x) in NSHDL, respectively]. Notably, these patients failed to improve with topical cholesterol alone, but both responded to dual treatment with cholesterol plus lovastatin.Citation54 Both excessive scale and epidermal hyperplasia diminished greatly, and one patient displayed improved mobility of underlying extremities by 6–8 weeks of treatment. In principal, this approach bridges the two poles of symptomatic vs. curative (i.e., gene) therapy. It also displays the following inherent advantages: (1) it is disease-targeted and mechanism-based specificity; (2) it is inherent safe; (3) it is relatively low-cost; and (4) most-importantly, it exploits the accessibility of skin to assess efficacy, as well as providing the opportunity to assess the mechanisms responsible for positive outcomes.
Yet, topical pathogenesis-based therapy is not curative, and even if successful, it would need to be utilized for the duration of the patient's lifetime. Moreover, despite knowledge of disease pathogenesis, it is possible that such mechanism-targeted therapies may not always be effective, if multiple downstream pathways contribute to disease pathogenesis. However, since these patients' skin displays higher than normal water permeability, both the inhibitors and lipid end-products should be readily bioavailable. If either the drug or the lipid end-product fails to be delivered (suggested by a failure to respond to either pathway product or pathway product ± inhibitor), it is possible that co-applications of inhibitor and lipid product together could interfere with absorption of one or both.
Finally and importantly, it is possible that there will be benefits for the extracutaneous features of one or more of these disorders. In the case of disorders of distal cholesterol metabolism, the transdermal approach has the potential to deliver statins and cholesterol to extracutaneous tissues prior to hepatic first-pass metabolism, offering the tantalizing prospect that topical therapy could improve the extracutaneous manifestations in one or more of these disorders. It is highly likely that the cutaneous phenotype reflects pathogenic mechanisms that also are on-going in extracutaneous tissues, successful pathogenesis-based therapy for the ichthyoses could point to comparable approach(es) to treat/prevent the extracutaneous manifestations of these disorders.
Abbreviations
| CHH | = | conradi-hünermann-happle syndrome |
| CHILD | = | congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome |
| CDPX2 | = | X-linked chondrodysplasia punctata type 2 |
| CSO4 | = | cholesterol sulfate |
| DHCR7 | = | 7-dehydrocholesterol reductase |
| EI | = | epidermolytic ichthyosis |
| EBP | = | emopamil-binding protein |
| HI | = | harlequin ichthyosis |
| IFAP | = | ichthyosis follicularis, patricia and photophobia syndrome |
| LB | = | lamellar bodies |
| L/NL | = | lamellar/non-lamellar |
| NS | = | netherton syndrome |
| SC | = | stratum corneum |
| SLOS | = | smith-lemli-opitz syndrome |
| SLS | = | sjögren-larsson syndrome |
| SSase | = | steroid sulfatase; XLI, X-linked ichthyosis |
Figures and Tables
Figure 1 Enzymatic stages in distal cholesterol metabolites and their associated clinical disorders. Syndromic disorders occur with mutations in 7 of the 9 post-lanosterol steps in late cholesterol synthesis (indicated by bold/italics). A prominent cutaneous phenotype (ichthyosis) occurs in 6 of these diseases (indicated by bold/italics & underline).
Figure 2 Pathogenesis-based therapy for disorders of distal cholesterol metabolites: Bases for improvement in clinical studies.
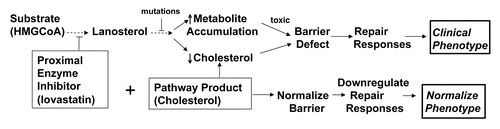
Table 1 Pathogenic mechanisms and potentially diagnostic features in disorders of distal cholesterol metabolism
Table 2 Pathogenesis and pathway-based therapy of inherited disorders of distal cholesterol metabolism
Acknowledgements
We are indebted to Joan Wakefield for superb editorial and graphic assistance. These studies were supported by NIH grant AR019098 and the Medical Research Service, Department of Veterans Affairs, San Francisco, CA.
References
- Moskowitz DG, Fowler AJ, Heyman MB, Cohen SP, Crumrine D, Elias PM, et al. Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function and energy expenditure. J Pediatr 2004; 145:82 - 92
- Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res 2008; 49:697 - 714
- Schmuth M, Gruber R, PM E, Williams M. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv Dermatol 2007; 23:231 - 256
- Elias PM, Feingold KR. Does the tail wag the dog? Role of the barrier in the pathogenesis of inflammatory dermatoses and therapeutic implications. Arch Dermatol 2001; 137:1079 - 1081
- Elias PM, Cullander C, Mauro T, Rassner U, Komuves L, Brown BE, et al. The secretory granular cell: the outermost granular cell as a specialized secretory cell. J Invest Dermatol Symp Proc 1998; 3:87 - 100
- Feingold KR. The outer frontier: the importance of lipid metabolism in the skin. J Lipid Res 2009; 50:417 - 422
- Elias P, Williams M, Crumrine D, Schmuth M. Ichthyoses—clinical, biochemical, pathogenic and diagnostic assessment. Curr Probl Dermatol 39:144
- Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res 2011; 52:6 - 34
- Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet 2001; 2:299 - 341
- Haas D, Kelley RI, Hoffmann GF. Inherited disorders of cholesterol biosynthesis. Neuropediatrics 2001; 32:113 - 122
- Moebius FF, Fitzky BU, Glossmann H. Genetic defects in postsqualene cholesterol biosynthesis. Trends Endocrinol Metab 2000; 11:106 - 114
- Herman GE. Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet 2003; 12:75 - 88
- Porter FD. Human malformation syndromes due to inborn errors of cholesterol synthesis. Curr Opin Pediatr 2003; 15:607 - 613
- FitzPatrick DR, Keeling JW, Evans MJ, Kan AE, Bell JE, Porteous ME, et al. Clinical phenotype of desmosterolosis. Am J Med Genet 1998; 75:145 - 152
- Krakowiak PA, Wassif CA, Kratz L, Cozma D, Kovarova M, Harris G, et al. Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol-5-desaturase deficiency. Hum Mol Genet 2003; 12:1631 - 1641
- He M, Kratz L, Michel lJ, Vallejo A, Ferris L, Kelley R, Hoover J, et al. Mutations in the SC4MOL gene encoding a novel methyl sterol oxidase cause psoriasiform dermatitis, microcephaly and developmental delay. Nature Precedings 2008; hdl:10101/npre.2008.2163.1
- Elias PM, Crumrine D, Rassner U, Hachem JP, Menon GK, Man W, et al. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol 2004; 122:314 - 319
- Singh P, Saxena R, Paila YD, Jafurulla M, Chattopadhyay A. Differential effects of cholesterol and desmosterol on the ligand binding function of the hippocampal serotonin(1A) receptor: implications in desmosterolosis. Biochim Biophys Acta 2009; 1788:2169 - 2173
- Rodriguez-Acebes S, de la Cueva P, Fernandez-Hernando C, Ferruelo AJ, Lasuncion MA, Rawson RB, et al. Desmosterol can replace cholesterol in sustaining cell proliferation and regulating the SREBP pathway in a sterol-Delta24-reductase-deficient cell line. Biochem J 2009; 420:305 - 315
- Feingold KR. The regulation and role of epidermal lipid synthesis. Adv Lipid Res 1991; 24:57 - 82
- Bjorkhem I. Are side-chain oxidized oxysterols regulators also in vivo?. J Lipid Res 2009; 50:213 - 218
- Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet 2003; 33:508 - 513
- Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr Suppl 2006; 95:16 - 23
- Clayton PT, Kalter DC, Atherton DJ, Besley GT, Broadhead DM. Peroxisomal enzyme deficiency in X-linked dominant Conradi-Hunermann syndrome. J Inherit Metab Dis 1989; 12:358 - 360
- Holmes RD, Wilson GN, Hajra AK. Peroxisomal enzyme deficiency in the Conradi-Hunerman form of chondrodysplasia punctata. N Engl J Med 1987; 316:1608
- Emami S, Rizzo WB, Hanley KP, Taylor JM, Goldyne ME, Williams ML. Peroxisomal abnormality in fibroblasts from involved skin of CHILD syndrome. Case study and review of peroxisomal disorders in relation to skin disease. Arch Dermatol 1992; 128:1213 - 1222
- Wilson CJ, Aftimos S. X-linked dominant chondrodysplasia punctata: a peroxisomal disorder?. Am J Med Genet 1998; 78:300 - 302
- Goldyne ME, Williams ML. CHILD syndrome. Phenotypic dichotomy in eicosanoid metabolism and proliferative rates among cultured dermal fibroblasts. J Clin Invest 1989; 84:357 - 360
- Emami S, Hanley KP, Esterly NB, Daniallinia N, Williams ML. X-linked dominant ichthyosis with peroxisomal deficiency. An ultrastructural and ultracytochemical study of the Conradi-Hunermann syndrome and its murine homologue, the bare patches mouse. Arch Dermatol 1994; 130:325 - 336
- Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta 2006; 1763:1733 - 1748
- DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res 2008; 18:609 - 621
- Yu H, Patel SB. Recent insights into the Smith-Lemli-Opitz syndrome. Clin Genet 2005; 68:383 - 391
- Bommannan D, Menon GK, Okuyama H, Elias PM, Guy RH. Sonophoresis II. Examination of the mechanism(s) of ultrasound-enhanced transdermal drug delivery. Pharm Res 1992; 9:1043 - 1047
- Ikegawa S, Ohashi H, Ogata T, Honda A, Tsukahara M, Kubo T, et al. Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata. Am J Med Genet 2000; 94:300 - 305
- Tsai JC, Guy RH, Thornfeldt CR, Gao WN, Feingold KR, Elias PM. Metabolic approaches to enhance transdermal drug delivery: Effect of lipid synthesis inhibitors. J Pharm Sci 1996; 85:643 - 648
- Tint GS, Yu H, Shang Q, Xu G, Patel SB. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J Lipid Res 2006; 47:1535 - 1541
- Marcos J, Shackleton CH, Buddhikot MM, Porter FD, Watson GL. Cholesterol biosynthesis from birth to adulthood in a mouse model for 7-dehydrosterol reductase deficiency (Smith-Lemli-Opitz syndrome). Steroids 2007; 72:802 - 808
- Andersson HC, Kratz L, Kelley R. Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet 2002; 113:315 - 319
- Happle R. Lyonization and the lines of Blaschko. Hum Genet 1985; 70:200 - 206
- Happle R. X-linked dominant chondrodysplasia punctata. Review of literature and report of a case. Hum Genet 1979; 53:65 - 73
- Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, et al. Mutations in a delta8delta7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet 1999; 22:286 - 290
- Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, et al. Mutations in the gene encoding 3beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet 1999; 22:291 - 294
- Grange DK, Kratz LE, Braverman NE, Kelley RI. CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet 2000; 90:328 - 335
- Kubilus J, Tarascio AJ, Baden HP. Steroid-sulfatase deficiency in sex-linked ichthyosis. Am J Hum Genet 1979; 31:50 - 53
- Epstein E Jr, Krauss RM, Shackleton CH. X-linked ichthyosis: increased blood cholesterol sulfate and electrophoretic mobility of low-density lipoprotein. Science 1981; 214:659 - 660
- Elias PM, Williams ML, Maloney ME, Bonifas JA, Brown BE, Grayson S, Epstein EHJ. Stratum corneum lipids in disorders of cornification. Steroid sulfatase and cholesterol sulfate in normal desquamation and the pathogenesis of recessive X-linked ichthyosis. J Clin Invest 1984; 74:1414 - 1421
- Bergner EA, Shapiro LJ. Metabolism of 3H-dehydroepiandrosterone sulphate by subjects with steroid sulphatase deficiency. J Inherit Metab Dis 1988; 11:403 - 415
- Williams ML, Elias PM. Stratum corneum lipids in disorders of cornification: increased cholesterol sulfate content of stratum corneum in recessive x-linked ichthyosis. J Clin Invest 1981; 68:1404 - 1410
- Long SA, Wertz PW, Strauss JS, Downing DT. Human stratum corneum polar lipids and desquamation. Arch Dermatol Res 1985; 277:284 - 287
- Ranasinghe AW, Wertz PW, Downing DT, Mackenzie IC. Lipid composition of cohesive and desquamated corneocytes from mouse ear skin. J Invest Dermatol 1986; 86:187 - 190
- Ponec M, Williams ML. Cholesterol sulfate uptake and outflux in cultured human keratinocytes. Arch Dermatol Res 1986; 279:32 - 36
- Zettersten E, Man MQ, Sato J, Denda M, Farrell A, Ghadially R, et al. Recessive x-linked ichthyosis: role of cholesterol-sulfate accumulation in the barrier abnormality. J Invest Dermatol 1998; 111:784 - 790
- Evers BM, Farooqi MS, Shelton JM, Richardson JA, Goldstein JL, Brown MS, Liang G. Hair growth defects in Insig-deficient mice caused by cholesterol precursor accumulation and reversed by simvastatin. J Invest Dermatol 2010; 130:1237 - 1248
- Paller A, Rodriguez-Martin M, Van Steensel M, Sorrell J, Heath C, Crumrine D, et al. Pathogenesis (pathway)-based therapy and genetic basis for lateralization in CHILD syndrome. SID 2011 Annual Meeting 2011; Phoenix AZ
- Correa-Cerro LS, Wassif CA, Kratz L, Miller GF, Munasinghe JP, Grinberg A, et al. Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum Mol Genet 2006; 15:839 - 851
- Mirza R, Hayasaka S, Takagishi Y, Kambe F, Ohmori S, Maki K, et al. DHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermis. J Invest Dermatol 2006; 126:638 - 647
- Mirza R, Qiao S, Murata Y, Seo H. Requirement of DHCR24 for postnatal development of epidermis and hair follicles in mice. Am J Dermatopathol 2009; 31:446 - 452
- Vahlquist A, Ganemo A, Virtanen M. Congenital ichthyosis: an overview of current and emerging therapies. Acta Derm Venereol 2008; 88:4 - 14
- Fritsch PO. Retinoids in psoriasis and disorders of keratinization. J Am Acad Dermatol 1992; 27:8 - 14
- Leachman SA, Hickerson RP, Schwartz ME, Bullough EE, Hutcherson SL, Boucher KM, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther 2010; 18:442 - 446
- Feingold KR, Man MQ, Proksch E, Menon GK, Brown BE, Elias PM. The lovastatin-treated rodent: a new model of barrier disruption and epidermal hyperplasia. J Invest Dermatol 1991; 96:201 - 209
- Menon GK, Feingold KR, Mao-Qiang M, Schaude M, Elias PM. Structural basis for the barrier abnormality following inhibition of HMG CoA reductase in murine epidermis. J Invest Dermatol 1992; 98:209 - 219