Abstract
Colorectal cancer (CRC) is the third most common cancer in the United States. Approximately 90% of colon cancer deaths arise from the metastasis of primary tumors. Aberrant expression of Wnt5a, one of the WNT signaling factors, has been reported during colon cancer development and progression. We found that both mRNA and protein expression of Wnt5a were decreased in the highly metastatic human colon cancer cell line SW620 compared with the non-metastatic human colon cancer cell SW480. This study tested the hypothesis that the silencing of Wnt5a in metastatic human colon cancer cells is related to altered epigenetic modifications. Wnt5a expression was not responsive to DNA methyltransferase inhibitor 5-aza-cytidine treatment. However, histone deacetylase (HDAC) inhibitors trichostatin A (TSA) and sodium butyrate (NaBt) significantly increased Wnt5a mRNA expression in SW620. Importantly, lower transcription of Wnt5a in SW620 than SW480 corresponded to multiple histone modifications, including lower levels of acetylated histone H3, H4 and H3K4me2 and higher levels of H3K27me3 in the promoter region. The increase of H3Ac, H4Ac and H3K4me2 after NaBt treatment in SW620 confirmed the involvement of histone modifications in the transcriptional regulation of Wnt5a. Additionally, NaBt treatment increased β-catenin signaling and diminished the difference in cell adhesion ability between non-metastatic SW480 and metastatic SW620, suggesting that the HDAC inhibitor plays critical roles in the WNT signaling pathway and cell physiology that relate to metastasis. In conclusion, our study suggests the importance of Wnt5a in colon cancer metastasis and also indicates that Wnt5a silencing in the highly invasive human colon cancer cell line might result from transcriptional regulation of the gene by histone modifications.
Introduction
Colorectal cancer (CRC) is the third most common and the third death-leading cancer in the United States.Citation1 It accounts for 9–10% of deaths from cancer.Citation1 Despite the recent and continuous improvements in diagnosis and treatments, more than 50% of CRC metastasize to liver, lung and lymph nodes.Citation2 The five-year survival rate remains below 50% for patients with metastatic CRC. Thus, there is an urgent need to elucidate the mechanism of CRC metastasis.
Wnt5a is one of the glycoproteins in the Wnt family of signaling molecules. It has been shown to stimulate the intracellular Ca2+ release and activation of PKC (i.e., non-canonical Wnt pathwayCitation3,Citation4). In addition, Wnt5a is involved in the canonical WNT signaling pathway (i.e., β-catenin-mediated pathway) by either activating or antagonizing it.Citation3,Citation5-Citation7 These diverse characteristics may be attributed to the tumor-suppressing or tumor-stimulating effects of Wnt5a, depending on cancer types and stages.Citation8-Citation11 For example, upregulation of Wnt5a is associated with breast cancer,Citation12 prostate cancerCitation8 and melanoma,Citation9 suggesting its oncogene role in these cancers. On the other hand, it shows tumor-suppressing effects in colorectal cancer,Citation10 thyroid cancerCitation11 and acute lymphoblastic leukemia.Citation13 Specifically in colon cancer, Dejmek et al. reported that Wnt5a protein expression was reduced in Dukes B colon carcinomas compared with normal colon tissue.Citation14 They also detected that Wnt5a expression was negatively correlated with 5-year survival of patients, suggesting the loss of Wnt5a as a potential prognostic marker for colon cancer progression.Citation10
The Wnt5a protein has been shown to regulate cell adhesion, motility and polarity.Citation14,Citation15 Knockdown of Wnt5a impaired cell adhesion and spreading, indicating its important role in cell adhesion.Citation16 β-catenin/E-cadherin-mediated cell-cell adhesion was reduced by loss of Wnt5a in breast carcinomas, suggesting Wnt5a is a good prognostic factor for longer disease-free survival in human breast cancer.Citation12 In colon cancer, Wnt5a inhibited cell proliferation and impaired cell migration, a function that is necessary for tumor invasion and metastasis.Citation11,Citation14 The function of Wnt5a in colon cancer suppression suggests its potential for inhibiting CRC metastasis.
It has been shown that epigenetic regulation, mainly DNA methylation and histone modifications, play important roles in the expression of Wnt5a. Ying et al.Citation10 observed silencing of Wnt5a in most CRC cell lines due to promoter methylation. Jensen et al.Citation17 reported that arsenic exposure in human bladder cancer cells induced histone modifications in correspondence with the transcriptional activation of Wnt5a. However, the relationship between Wnt5a expression and epigenetic modifications in CRC metastasis remains undefined.
To understand the epigenetic regulation of Wnt5a in the progression of colon cancer metastasis, we used two human colon cancer cell lines, SW480 and SW620, which have been established from the same colon cancer patient. SW480 was derived from a colon carcinoma in the 50-y-old male patient, while SW620 was derived from the lymph node metastases in the same patient one year later.Citation18 Although these two cell lines are isogenic, they differ remarkably in many cell properties, such as cell morphology, tumorigenicity, adhesion, migration capacity and metastatic potential.Citation19-Citation21 The unique properties of SW480 and SW620 make them a good model for studying epigenetic modifications in the late stages of colon cancer progression.
In the current study, we aimed to test the hypothesis that CRC metastasis is associated with aberrant expression of Wnt5a, which is regulated by epigenetic mechanisms. Repression of Wnt5a in SW620 corresponded to histone modifications and was partially reversed by HDAC inhibitors. HDAC inhibitor also modified β-catenin and enhanced the adhesion ability of SW620. Taken together, our findings indicate that histone modifications play important roles in CRC metastasis by regulating Wnt5a signaling.
Results
Wnt5a was silenced in the highly metastatic cell line SW620
To compare the mRNA expression of Wnt5a in human colon cancers with different invasiveness, real-time PCR analysis was performed in colon cancer cell line SW480, which was established from the primary tumor site and in the highly metastatic cell line SW620, which was established from a lymph node metastasis from the same patient a year later. The level of Wnt5a mRNA in SW480 was approximately 1,000 times higher than in SW620 (p < 0.05, ).
Figure 1. Protein and mRNA expression of Wnt5a in non-metastatic human colon cancer cell line SW480 and highly metastatic cell line SW620. (A) Relative mRNA expression of Wnt5a in SW480 and SW620 presented as the ratio to the housekeeping gene L7a (n = 3). mRNA was analyzed using real-time RT-PCR. The values are presented as the mean ± SEM *p < 0.05 when comparing SW620 to SW480. (B) Wnt5a protein expression in SW480 and SW620 using immunofluorescent staining. Wnt5a protein was analyzed after immunofluorescent staining using an antibody against Wnt5a and an Alexa Fluor 647-labeled secondary antibody (red). The coverslips were also counterstained with Hoechst 33342 fluorescent stain to detect the nucleus (blue). The two pictures were overlaid to show Wnt5a staining on top of the nuclear staining (merged). (C) Quantification of immunofluorescent staining of Wnt5a (stained in red) in SW480 and SW620. Over 20 cells per slide were randomly chosen for quantification. Data was shown as relative intensity per pixel based on AxioVision4.7 software. The values are presented as the mean ± SEM, *p < 0.05 when comparing SW620 to SW480.
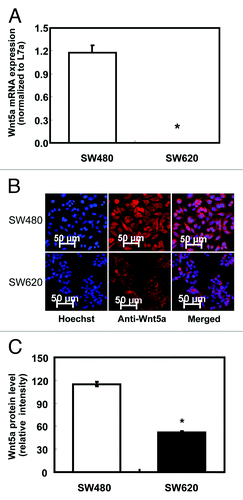
We also tested Wnt5a protein expression by immunofluorescent staining (, anti-Wnt5a). Quantitative analysis of immunofluorescent staining indicated that Wnt5a protein was significantly less expressed in SW620 compared with SW480 (p < 0.05, ), which was consistent with the mRNA expression analysis by real-time PCR.
Histone deacetylase inhibitors, but not a DNA methylation inhibitor, increased Wnt5a expression in SW620
There are two putative CpG islands in the promoter region of human Wnt5a gene (from -1000 to exon 1, as predicted by www.urogene.org/methprimer), indicating the possible role of DNA methylation in the regulation of Wnt5a gene expression in human. To examine the involvement of DNA methylation in the silencing of Wnt5a in SW620, we treated SW620 with 4 and 8 µmol/L of the DNA methylation inhibitor 5-aza-cytidine. The mRNA expression of Wnt5a did not change after 5-aza-cytidine treatment (), which suggested that the decreased expression of Wnt5a in SW620 was not directly caused by DNA methylation.
Figure 2. Expression of Wnt5a mRNA after 5-aza-cytidine (Azacitidine), trichostatin A (TSA) and sodium butyrate (NaBt) treatment. (A) Relative Wnt5a mRNA level was tested in SW480 and SW620 treated with Azacitidine (0, 4 and 8 μmol/L) for 48 h. mRNA was analyzed using real-time RT-PCR and presented as the ratio to L7a (n = 3). (B) Wnt5a mRNA level was tested in SW620 treated with TSA (50 and100 nmol/L) or NaBt (2.5 and 5 mmol/L) for 48 h. mRNA was analyzed using real-time PCR and presented as the ratio to L7a (n = 3). The values are presented as the mean ± SEM, *p < 0.05 when compared with the DMSO-treated SW620 control group.
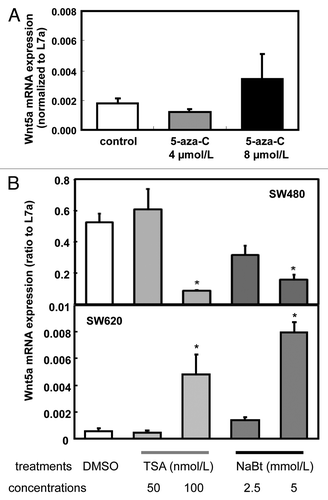
To test whether histone acetylation contributes to altered expression of Wnt5a during colon cancer metastasis, we exposed the cells to HDAC inhibitors TSA (50 or 100 nmol/L) and NaBt (2.5 or 5 mmol/L). At the higher concentration, both TSA and NaBt increased the expression of Wnt5a mRNA in SW620 but reduced it in SW480 cells (). mRNA expression of Wnt5a was upregulated ~8 times by 100 nmol/L of TSA and ~14 times by 5 mmol/L of NaBt in SW620 (p < 0.05, ). This observation indicates that HDAC was involved in the regulation of Wnt5a expression in a cell line that models the development of colon cancer metastasis.
Histone modifications were associated with the regulation of Wnt5a expression in SW480 and SW620
To further test our hypothesis that histone modifications are involved in regulating Wnt5a expression in colon cancer cells, we used ChIP to investigate the chromatin structure at the promoter region (, upper panel) and coding region (, lower panel) of the gene. When comparing SW480 and SW620, we observed much lower levels of H3Ac, H4Ac and H3K4me2 in SW620, whereas H3K27me3 was higher in SW620 than in SW480. Moreover, the promoter region and coding region showed similar patterns in terms of chromatin structure, even though they are 6 kb apart ()
Figure 3. ChIP analysis of chromatin modifications in SW480 and SW620. (A) Histone modifications at the promoter region (-96/-22, upper panel) and coding region (+6424/+6495, lower panel) of human Wnt5a in SW480 and SW620 (n = 3). Data are normalized to the input DNA. The values are presented as the mean ± SEM *p < 0.05 indicates statistical significance compared with SW480. (B) Histone modifications at the promoter region (-96/-22, upper panel) and coding region (+6424/+6495, lower panel) of human Wnt5a in SW620 after NaBt treatment (5 mmol/L) for 48 h (n = 3). Data are normalized to the input DNA. The values are presented as the mean ± SEM *p < 0.05 when comparing to the non-treated SW620 control group.
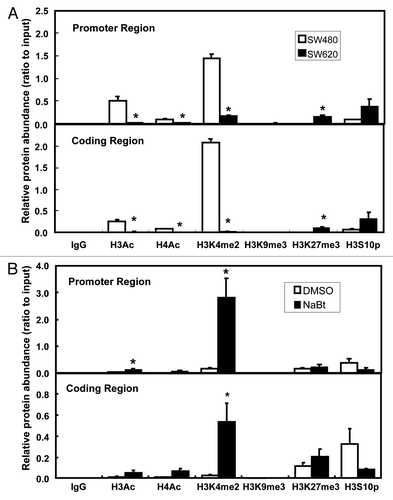
ChIP was also performed in SW620 following NaBt (5 mmol/L) treatment. After the 48 h treatment, H3Ac was significantly increased at the promoter region (p < 0.05, , upper panel), but not at the coding region (, lower panel). NaBt treatment also significantly induced H3K4me2 in both the promoter and the coding regions of Wnt5a in SW620 (p < 0.05, ). Moreover, under NaBt treatment, the level of H3K4me2 was ~4 times higher at the promoter region than at the coding region (p < 0.05, ).
HDAC inhibitor upregulated the WNT signaling pathway in SW620
To examine the impact of HDAC inhibitor on the WNT signaling pathway, we examined the NaBt-treated SW620 cells for the expression of β-catenin, the key component of WNT signaling, by immunofluorescent staining (). Quantitative analysis showed that β-catenin protein content was much lower in SW620 than in SW480 (p < 0.05, ). NaBt treatment restored/increased β-catenin levels in SW620 to levels even higher than those in SW480 (p < 0.05, ). This observation suggests that the expression of Wnt5a in these human colon cancer cell lines is positively associated with the activation of β-catenin.
Figure 4. Influence of NaBt treatment on the WNT signaling pathway and cell adhesion ability of SW620. (A) Expression of β-catenin protein in SW480, SW620 and SW620 treated with NaBt (5 mmol/L) for 48 h using immunofluorescence. β-catenin protein was analyzed by immunofluorescent staining using an antibody against β-catenin and an Alexa Fluor 647-labeled secondary antibody (red). The coverslips were also counterstained with Hoechst 33342 fluorescent stain to detect the nucleus (blue). The two pictures were overlaid to show β-catenin staining on top of the nuclear staining (merged). (B) Quantification of immunofluorescent staining of β-catenin protein (stained in red) in SW480, SW620 and SW620 treated with NaBt. Over 20 cells per slide were randomly chosen for quantification. Data was shown as relative intensity per pixel based on AxioVision4.7 software. The values are presented as the mean ± SEM. Values with different letters are statistically different (p < 0.05). (C) Cell adhesion ability of SW480, SW620 and SW620 treated with NaBt (5 mmol/L) for 48 h (n = 3). Cells grown on fibronectin-coated plates were fixed and stained with crystal violet. The numbers of adherent cells were estimated by absorbance reading at 562 nm. The values are presented as the mean ± SEM. Values with different letters are statistically different (p < 0.05).
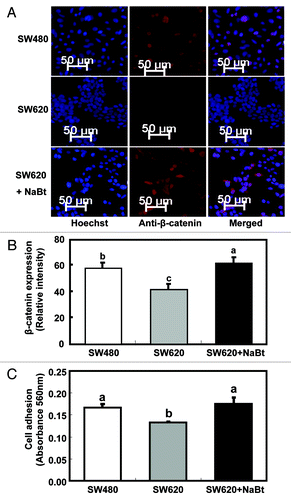
HDAC inhibitor enhanced adhesion ability of SW620 to a level similar to that in SW480
An in vitro cell adhesion assay was performed to investigate the impact of Wnt5a on cell adhesion, which is related to cell interactions and cancer metastasis (). The adhesion ability of SW620 was lower than that of SW480 (p < 0.05). Importantly, NaBt treatment elevated the cell adhesion ability of SW620, diminishing the difference between the two cell lines.
Discussion
In this study, mechanistic analysis was conducted to determine the relationship between colon cancer metastasis and silencing of Wnt5a. Highly metastatic colon cancer cell line SW620 showed much lower expression of Wnt5a as well as lower activity of the WNT signaling pathway than primary colon cancer cell line SW480. More importantly, restoration of Wnt5a expression in SW620 by HDAC inhibitors, and the corresponding changes in chromatin modifications, indicates that silencing of the Wnt5a gene in this cell line was mainly due to histone modifications that occurred at the Wnt5a gene regions.
It has become increasingly evident that epigenetic changes contribute to cancer progression and metastasis.Citation22-Citation24 HDAC was responsible for the suppression of E-cadherin in metastatic pancreatic cancer and thus induced the epithelial to mesenchymal transition.Citation25 Epigenetic downregulation of secreted frizzled-related protein 5 (SFRP5), an antagonist of the WNT signaling pathway, lead to cancer progression.Citation26,Citation27 Moreover, our group previously reported that the metastasis suppressor NDRG1 was transcriptionally downregulated by epigenetic alterations in CRC metastasis.Citation21 The present study showed that the epigenetic silencing of Wnt5a, a WNT signaling molecule, was involved in CRC metastasis.
Histone modifications, such as acetylation, methylation and phosphorylation, influence gene expression by altering chromatin environments.Citation28 The present study revealed that multiple histone modifications were associated with the aberrant expression of Wnt5a in metastatic colon cancer cell line SW620. Silencing of Wnt5a in SW620 was positively correlated with low levels of active histone modifications, including the acetylation of histone H3 and H4, and the methylation of histone H3 lysine 4 at its promoter. We also tested the levels of the repressive histone marker H3K27me3, whose enrichment correlates with gene repression not only in normal developmentCitation29 but also in cancers.Citation30,Citation31 Moreover, the silencing of tumor-suppressor genes in cancer regulated by H3K27me3 has been shown to be potentially independent of promoter DNA methylation.Citation30 In the present study, we observed much higher levels of H3K27me3 in SW620 than in SW480, which was consistent with the lower expression of Wnt5a in SW620. H3K9me3 was previously implicated in gene silencing.Citation32 However, H3K9me3 in both SW480 and SW620 was nearly undetectable, suggesting that methylation of H3K9 did not influence Wnt5a expression in this case. Apart from histone acetylation and methylation, histone phosphorylation may also affect the expression of Wnt5a. Phosphorylated H3S10 plays a critical role in the dynamic condensation/decondensation during the cell cycle.Citation33 Changes in chromatin structure induced by phosphorylation of histone H3S10 cause other histone modifications, either active or repressive, that interact and further regulate transcription.Citation34 The present study indicates that increased phosphorylation of histone H3S10 may be associated with the silencing of the Wnt5a gene in SW620.
The status of histone acetylation in SW480 and SW620 were consistent with our finding that Wnt5a mRNA was significantly increased by treatment with HDAC inhibitors TSA and NaBt, which belong to two different group types of HDAC inhibitors.Citation35 Both TSA and NaBt induced Wnt5a expression in a concentration-dependent manner. To specifically test the impact of HDAC inhibitors on the histone landscape, ChIP was performed in SW620 following NaBt treatment. Interestingly, both acetylated histone H3 and methylated H3 lysine 4 were significantly increased in the promoter region by NaBt induction. A similar observation was reported by Mossman et al.,Citation36 leading to the conclusion that transcriptional reactivation of epigenetically-silenced genes in colon cancer cells requires increased H3 acetylation and H3 methylation.
Our study also indicates that the HDAC inhibitor altered the WNT signaling pathway and caused metastasis-related physiological changes. As the key component of canonical WNT signaling, β-catenin acts as the marker of the activation of the pathway. Although Wnt5a activates β-catenin-independent signaling and was mostly considered as the antagonist of the β-catenin-dependent WNT pathway,Citation37 several studies showed that overexpression of Wnt5a and its receptors activated the β-catenin pathway.Citation5,Citation6 Our finding suggested that the upregulation of Wnt5a by NaBt exposure induced the activation of β-catenin signaling. Furthermore, NaBt exposure increased the cell adhesion ability of SW620 to a level similar to that of SW480, indicating the impact of HDAC inhibition on the physiology of metastatic colon cancer cells. The change in cell adhesion ability may be attributed to the increased Wnt5a expression by HDAC inhibitor, as it was shown that knockdown of Wnt5a impaired cell adhesion and spreading.Citation16 Further research needs to be conducted to investigate the role that epigenetic mechanisms of histone modifications may have in Wnt5a gene expression, and these future findings may lead to epigenetic therapies for colon cancer metastasis.
Several studies have shown that Wnt5a expression was regulated by DNA methylation.Citation38-Citation40 Our current finding that Wnt5a was silenced in SW620 but not in SW480 was consistent with the report by Ying et al., showing that Wnt5a was silenced in four out of six human colon cancer cell lines.Citation10 They indicated the possible correlation of downregulation of Wnt5a in SW620 with the high methylation status at its promoter, as tested by methylation-specific PCR. In the current study, we did not detect induction of Wnt5a expression after 5-aza-cytidine treatment in SW620, indicating the lack of response to DNA demethylation in this cell line. Therefore, at least in SW620 cells, hypermethylation of the Wnt5a promoter may be a consequence of a generally high DNA methylation environment and it is not directly linked to the transcriptional regulation of Wnt5a. Considering the complexity of epigenetic modifications in colon cancer progression, future research is critically needed to study the interactions of different epigenomic components, mainly DNA methylation and histone/chromatin modifications.
Taken together, this study provides insights into the epigenetic modifications of Wnt5a, a critical mediator of the WNT signaling pathway, which was decreased during CRC metastasis. Our study also provides more evidence in terms of the general effects of HDAC inhibitors on the commonly aberrant WNT signaling pathway in advanced CRC stages. Our findings advance the understanding of molecular mechanisms of colon cancer metastasis, especially at the level of epigenetic remodeling.
Materials and Methods
Cell culture
The human colon cancer cell lines SW480 and SW620 were purchased from ATCC. SW480 and SW620 are isogenic cell lines established from the colon cancer of the same patient.Citation18 SW480 was the primary colon cancer derived from the patient and SW620 was derived from the lymph node metastasis from the same person one year later. Minimum essential medium (MEM) was purchased from the Cell Media Facility at the University of Illinois. Unless otherwise mentioned, all general chemicals were obtained from Fisher Scientific (www.fishersci.com). Cell culture ware was purchased from Sarstedt. Both cell lines were cultured in MEM supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic solution (ABAM) at 37°C in a humid incubator with 5% CO2. All experiments were performed using cells with 2–6 passages. Cell line verification was routinely performed in the laboratory according to the guidelines by ATCC (Technical Bulletin No. 8, www.atcc.org).
5-aza-cytidine, TSA and NaBt treatment
SW480 and SW620 cells were plated at 0.3 × 106 per 60 mm culture dish in regular growth media. After overnight incubation, cells were treated with culture media containing 0, 4 or 8 µmol/L 5-aza-cytidine (Azacitidine, Sigma-Aldrich), 50 or 100 nmol/L TSA (Sigma), or 2.5 or 5 mmol/L NaBt for 2 d. Treatment media were refreshed after 1 d. Total RNAs were isolated after each treatment and mRNA expression was analyzed by real time PCR as described below.
Quantitative real-time PCR
Cells were harvested in TriReagent (Sigma-Aldrich). Total RNAs were isolated following the manufacturer’s instructions. cDNA was synthesized from total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNA was then analyzed by two-step real-time PCR using the 7300 real-time PCR system (Applied Biosystems) and detected with SYBR Green. In each reaction, 25 ng of synthesized cDNA were used in a 20 µL volume containing 10 µL Perfecta SYBR Green fast master mix (2x, Quanta BioSciences, www.vwr.com) and 0.25 µmol/L of each primer. Real-time PCR was performed using the following program: 95°C for 10 min, 40 cycles of 95°C for 15 sec followed by 60°C for 1 min. A serial dilution of a cDNA sample was used in the same PCR reaction to generate a standard curve, based on which the quantity of the unknown samples was calculated. Standard curves with slope of -3.3 ± 0.2 and R2 ≥ 0.98 were accepted. L7a was used as an internal control to normalize obtained raw data. PCR primers were synthesized by Integrated DNA Technologies (www.idtdna.com). The sequences are as follows: Wnt5a (+262/+340), sense 5'- GACCACATGCAGTACATCGGAGAAG -3' and antisense 5'-TCCACCCTCGATG TCGGAATTG-3'; L7a (+145/+208), sense 5'-TTTGG CATTGGACAGGACATCC-3' and antisense 5'-AGCGGGGCCATTT CACAAAG-3'.
Immunofluorescent staining
Cells were seeded and grown on coverslips in 6-well plates pre-coated with fibronectin. After 2 d incubation, cells were washed three times with PBS, fixed with 4% paraformaldehyde at room temperature for 30 min and washed with PBS. Fixed cells were then incubated in PBS containing 0.1% Triton X-100 at room temperature for 15 min and washed with PBS. Coverslips with cells were transferred onto slides and blocked with Image-iT™ FX Signal Enhancer (I36933, Invitrogen) for 30 min at room temperature. Slides were then incubated in PBS containing primary antibodies (1:150 dilution) at 37°C for 2 h, followed by 3x PBS washing, and then incubated with Alexa Fluor 647 goat anti-rabbit IgG (1:200, A-21245, Invitrogen) for 45 min in the dark. Nuclei were stained with 1 µmol/L Hoechst 33342 (H3570, Invitrogen) for 15 min in the dark. Approximately 120 µL ProLong Gold Antifade Reagent (P36934, Invitrogen) was added to PBS-washed and drained slide. Slides were kept in the dark at room temperature for 24 h for the ProLong Gold to cure and stored at 4°C until analysis. Images were acquired by fluorescence microscope (Zeiss Axiovert 200M with the Apotome). The primary antibodies used are as follows: Wnt5a, sc-30224 (Santa Cruz) and β-catenin, 9587 (cell signaling). The quantification of immunofluorescent staining was performed by AxioVision 4.7 software (Zeiss) after image acquisition. More than 20 cells (stained in red) in each slide were randomly chosen and the outline of each cell was drawn. The intensity per pixel of each selected cell was automatically generated by the software. Finally, the relative intensity per pixel in each group was calculated.
Chromatin immunoprecipitation (ChIP)
To determine the specific histone modifications related to the regulation of Wnt5a gene expression, ChIP analysis was performed according to a published protocol.Citation41 Approximately 15 × 106 cells per 150 mm dish were plated. Three dishes of cells were used for one treatment and sampling. After treatment, cells were cross-linked with 1% formaldehyde for 10 min on a rotator at room temperature. Cell pellets were resuspended in nuclei swelling buffer (5 mmol/L pipes pH 8.0, 85 mmol/L KCl, 0.5% NP40) and lysed in SDS lysis buffer (50 mmol/L TRIS-HCl pH 8.1, 10 mmol/L EDTA, 1% SDS) containing protease inhibitors and phosphatase inhibitors. The chromatin was sonicated on ice using Sonic Dismembrator (model F100, Fisher Scientific) with power set at 5 for 5 bursts of 40 sec each. Sheared chromatin was diluted with ChIP dilution buffer, aliquoted to 1 mL per antibody and incubated overnight with 2 µg of antibody/mL. The chromatin-antibody complex was precipitated with 60 µL of 50% (v/v) pre-blocked protein G-agarose slurry (Millipore). Normal rabbit IgG was used as the negative control. Supernatant from the normal rabbit IgG was saved as the input for total genomic DNA. Agarose with bound antibody was washed sequentially with 1 mL of the following solutions: low salt (20 mmol/L pH 8.0 TRIS-HCl, 0.1% SDS, 2 mmol/L EDTA, 150 mmol/L NaCl, 1% triton X-100), high salt (20 mmol/L pH 8.0 TRIS-HCl, 0.1% SDS, 2 mmol/L EDTA, 500 mmol/L NaCl, 1% triton X-100), LiCl (10 mmol/L pH 8.0 TRIS-HCl, 0.25 mmol/L LiCl, 1% NP40, 1 mmol/L EDTA, 1% sodium deoxycholate), and TE (pH 8.0). Chromatin complexes were eluted twice with 250 µL of the elution buffer (1% SDS and 50 mmol/L NaHCO3) at 37°C for 15 min and reversed-crosslinked with 0.2 mol/L NaCl at 65°C for 5 h. Chromatin DNA was purified using QiaPrep miniprep kit (Qiagen) after proteinase K digestion. Immunoprecipitated DNA was detected by real time PCR using primers targeting different regions of the Wnt5a gene: promoter region (-96/-22), sense 5'-GGTCTTTTGCACAATCACGCC-3' and antisense 5'-TTTCCAACGTCCATCAGCGAC-3'; coding region (+6424/+6495), sense 5'-GATGGCTGGAAGTGCAATGTCT-3' and antisense 5'-ACCTGGGCGAAGGAGAAAAA-3'. The antibodies used are as follows: Acetylated histone H3 (H3Ac, 06–599, Millipore), acetylated histone H4 (H4Ac, 06–866, Millipore), dimethyl-histone H3 lysine 4 (H3K4me2, 07–030, Millipore), trimethyl-histone H3 lysine 9 (H3K9me3, 07–442, Millipore), trimethyl-histone H3 lysine 27 (H3K27me3, 07–449, Millipore), normal rabbit IgG (IgG, sc-2027, Santa Cruz Biotechnologies), phosphorylated histone H3 serine 10 (H3S10P, sc-8656R, Santa Cruz).
Adhesion assay
Fibronectin-coated 96-well plates were used for the cell adhesion assay. For coating, plates were incubated with 0.3 µg fibronectin at 37°C for 1 h. Plates were blocked with blocking buffer (0.5% BSA in MEM) at 37°C for 1 h. Then, 0.02 × 106 cells were plated and incubated in a cell culture incubator for 30 min. Cells were fixed with 4% paraformaldehyde and then stained with crystal violet (5 mg/mL in 2% ethanol) for 10 min and washed with water. Fifty µL of 2% SDS was added to air-dried wells and incubated at room temperature for 30 min. The plate was read at 562 nm in a microplate reader EL × 800 (BioTek). Values of absorbance obtained at wavelength 562 nm represented relative amount of adherent cells.
Statistical analysis
Unpaired two-tailed Student t-test was performed for mRNA expression. One-way ANOVA using LSMeans was performed for ChIP results (SAS Institute Inc.). One-way ANOVA using tukey’s test was performed for adhesion results and immunofluorescent staining quantification. Data are presented as the mean ± Standard error of the means (SEM). The statistical significance was set at p < 0.05.
| Abbreviations: | ||
| CRC | = | colorectal cancer |
| HDAC | = | histone deacetylase |
| TSA | = | trichostatin A |
| NaBt | = | sodium butyrate |
| H3Ac | = | acetylated histone H3 |
| H4Ac | = | acetylated histone H4 |
| H3K4me2 | = | dimethyl histone H3 lysine 4 |
| H3K9me3 | = | trimethyl histone H3 lysine 9 |
| H3K27me3 | = | trimethyl histone H3 lysine 27 |
| H3S10P | = | phosphorylated histone H3 serine 10 |
Acknowledgments
The authors wish to thank other members of Pan and Chen laboratories at the University of Illinois at Urbana-Champaign for their technical advice and assistance. The authors acknowledge Dr. Rita Strakovsky for English copyediting. The authors wish to acknowledge the financial support by the Arnold O. Beckman Award from the Research Board at the University of Illinois at Urbana-Champaign (to H.C.). This study was also supported by a pilot grant from BRC/UIUC by the National Center for Complementary and Alternative Medicines (NCCAM), the Office of Dietary Supplements (ODS) and the National Cancer Institute (NCI, Grant Number P50AT006268 to H.C.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NCCAM, ODS, NCI, or the National Institutes of Health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010; 60:277 - 300; http://dx.doi.org/10.3322/caac.20073; PMID: 20610543
- Segal NH, Saltz LB. Evolving treatment of advanced colon cancer. Annu Rev Med 2009; 60:207 - 19; http://dx.doi.org/10.1146/annurev.med.60.041807.132435; PMID: 19630571
- Sheldahl LC, Park M, Malbon CC, Moon RT. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr Biol 1999; 9:695 - 8; http://dx.doi.org/10.1016/S0960-9822(99)80310-8; PMID: 10395542
- Slusarski DC, Yang-Snyder J, Busa WB, Moon RT. Modulation of embryonic intracellular Ca2+ signaling by Wnt-5A. Dev Biol 1997; 182:114 - 20; http://dx.doi.org/10.1006/dbio.1996.8463; PMID: 9073455
- Umbhauer M, Djiane A, Goisset C, Penzo-Méndez A, Riou JF, Boucaut JC, et al. The C-terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/beta-catenin signalling. EMBO J 2000; 19:4944 - 54; http://dx.doi.org/10.1093/emboj/19.18.4944; PMID: 10990458
- He X, Saint-Jeannet JP, Wang Y, Nathans J, Dawid I, Varmus H. A member of the Frizzled protein family mediating axis induction by Wnt-5A. Science 1997; 275:1652 - 4; http://dx.doi.org/10.1126/science.275.5306.1652; PMID: 9054360
- Olson DJ, Gibo DM. Antisense wnt-5a mimics wnt-1-mediated C57MG mammary epithelial cell transformation. Exp Cell Res 1998; 241:134 - 41; http://dx.doi.org/10.1006/excr.1998.4030; PMID: 9633521
- Yamamoto H, Oue N, Sato A, Hasegawa Y, Yamamoto H, Matsubara A, et al. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 2010; 29:2036 - 46; http://dx.doi.org/10.1038/onc.2009.496; PMID: 20101234
- Da Forno PD, Pringle JH, Hutchinson P, Osborn J, Huang Q, Potter L, et al. WNT5A expression increases during melanoma progression and correlates with outcome. Clin Cancer Res 2008; 14:5825 - 32; http://dx.doi.org/10.1158/1078-0432.CCR-07-5104; PMID: 18794093
- Ying J, Li H, Yu J, Ng KM, Poon FF, Wong SC, et al. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res 2008; 14:55 - 61; http://dx.doi.org/10.1158/1078-0432.CCR-07-1644; PMID: 18172252
- Kremenevskaja N, von Wasielewski R, Rao AS, Schöfl C, Andersson T, Brabant G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 2005; 24:2144 - 54; http://dx.doi.org/10.1038/sj.onc.1208370; PMID: 15735754
- Medrek C, Landberg G, Andersson T, Leandersson K. Wnt-5a-CKIalpha signaling promotes beta-catenin/E-cadherin complex formation and intercellular adhesion in human breast epithelial cells. J Biol Chem 2009; 284:10968 - 79; http://dx.doi.org/10.1074/jbc.M804923200; PMID: 19244247
- Roman-Gomez J, Jimenez-Velasco A, Cordeu L, Vilas-Zornoza A, San Jose-Eneriz E, Garate L, et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur J Cancer 2007; 43:2736 - 46; http://dx.doi.org/10.1016/j.ejca.2007.10.004; PMID: 18032022
- Dejmek J, Dejmek A, Säfholm A, Sjölander A, Andersson T. Wnt-5a protein expression in primary dukes B colon cancers identifies a subgroup of patients with good prognosis. Cancer Res 2005; 65:9142 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-05-1710; PMID: 16230369
- Witze ES, Litman ES, Argast GM, Moon RT, Ahn NG. Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science 2008; 320:365 - 9; http://dx.doi.org/10.1126/science.1151250; PMID: 18420933
- Matsumoto S, Fumoto K, Okamoto T, Kaibuchi K, Kikuchi A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J 2010; 29:1192 - 204; http://dx.doi.org/10.1038/emboj.2010.26; PMID: 20224554
- Jensen TJ, Wozniak RJ, Eblin KE, Wnek SM, Gandolfi AJ, Futscher BW. Epigenetic mediated transcriptional activation of WNT5A participates in arsenical-associated malignant transformation. Toxicol Appl Pharmacol 2009; 235:39 - 46; http://dx.doi.org/10.1016/j.taap.2008.10.013; PMID: 19061910
- Leibovitz A, Stinson JC, McCombs WB 3rd, McCoy CE, Mazur KC, Mabry ND. Classification of human colorectal adenocarcinoma cell lines. Cancer Res 1976; 36:4562 - 9; PMID: 1000501
- Hewitt RE, McMarlin A, Kleiner D, Wersto R, Martin P, Tsokos MJ, et al. Validation of a model of colon cancer progression. J Pathol 2000; 192:446 - 54; http://dx.doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH775>3.0.CO;2-K; PMID: 11113861
- Hewitt RE, Brown KE, Corcoran M, Stetler-Stevenson WG. Increased expression of tissue inhibitor of metalloproteinases type 1 (TIMP-1) in a more tumourigenic colon cancer cell line. J Pathol 2000; 192:455 - 9; http://dx.doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH777>3.0.CO;2-E; PMID: 11113862
- Li Q, Chen H. Transcriptional silencing of N-Myc downstream-regulated gene 1 (NDRG1) in metastatic colon cancer cell line SW620. Clin Exp Metastasis 2011; 28:127 - 35; http://dx.doi.org/10.1007/s10585-010-9366-4; PMID: 21184144
- Rodenhiser DI. Epigenetic contributions to cancer metastasis. Clin Exp Metastasis 2009; 26:5 - 18; http://dx.doi.org/10.1007/s10585-008-9166-2; PMID: 18386135
- Chik F, Szyf M, Rabbani SA. Role of epigenetics in cancer initiation and progression. Adv Exp Med Biol 2011; 720:91 - 104; http://dx.doi.org/10.1007/978-1-4614-0254-1_8; PMID: 21901621
- Li Q, Chen H. Epigenetic modifications of metastasis suppressor genes in colon cancer metastasis. Epigenetics 2011; 6:849 - 52; http://dx.doi.org/10.4161/epi.6.7.16314; PMID: 21758003
- von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009; 137:361 - 71, 371, e1-5; http://dx.doi.org/10.1053/j.gastro.2009.04.004; PMID: 19362090
- Su HY, Lai HC, Lin YW, Liu CY, Chen CK, Chou YC, et al. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int J Cancer 2010; 127:555 - 67; http://dx.doi.org/10.1002/ijc.25083; PMID: 19957335
- Kawakami K, Yamamura S, Hirata H, Ueno K, Saini S, Majid S, et al. Secreted frizzled-related protein-5 is epigenetically downregulated and functions as a tumor suppressor in kidney cancer. Int J Cancer 2011; 128:541 - 50; http://dx.doi.org/10.1002/ijc.25357; PMID: 20340127
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006; 441:349 - 53; http://dx.doi.org/10.1038/nature04733; PMID: 16625203
- Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 2008; 40:741 - 50; http://dx.doi.org/10.1038/ng.159; PMID: 18488029
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007; 39:232 - 6; http://dx.doi.org/10.1038/ng1950; PMID: 17200670
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001; 410:120 - 4; http://dx.doi.org/10.1038/35065138; PMID: 11242054
- Prigent C, Dimitrov S. Phosphorylation of serine 10 in histone H3, what for?. J Cell Sci 2003; 116:3677 - 85; http://dx.doi.org/10.1242/jcs.00735; PMID: 12917355
- Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet 2004; 20:214 - 20; http://dx.doi.org/10.1016/j.tig.2004.02.007; PMID: 15041176
- Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res 2011; 3:166 - 79; PMID: 21416059
- Mossman D, Scott RJ. Long term transcriptional reactivation of epigenetically silenced genes in colorectal cancer cells requires DNA hypomethylation and histone acetylation. PLoS One 2011; 6:e23127; http://dx.doi.org/10.1371/journal.pone.0023127; PMID: 21829702
- Kikuchi A, Yamamoto H, Sato A, Matsumoto S. Wnt5a: its signalling, functions and implication in diseases. Acta Physiol (Oxf) 2012; 204:17 - 33; http://dx.doi.org/10.1111/j.1748-1716.2011.02294.x; PMID: 21518267
- Li J, Ying J, Fan Y, Wu L, Ying Y, Chan AT, et al. WNT5A antagonizes WNT/β-catenin signaling and is frequently silenced by promoter CpG methylation in esophageal squamous cell carcinoma. Cancer Biol Ther 2010; 10:617 - 24; http://dx.doi.org/10.4161/cbt.10.6.12609; PMID: 20603606
- Wang Z, Chen H. Genistein increases gene expression by demethylation of WNT5a promoter in colon cancer cell line SW1116. Anticancer Res 2010; 30:4537 - 45; PMID: 21115903
- Roman-Gomez J, Jimenez-Velasco A, Cordeu L, Vilas-Zornoza A, San Jose-Eneriz E, Garate L, et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur J Cancer 2007; 43:2736 - 46; http://dx.doi.org/10.1016/j.ejca.2007.10.004; PMID: 18032022
- Chen H, Pan YX, Dudenhausen EE, Kilberg MS. Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient-responsive basic region/leucine zipper transcription factors as well as localized histone acetylation. J Biol Chem 2004; 279:50829 - 39; http://dx.doi.org/10.1074/jbc.M409173200; PMID: 15385533