Abstract
Assessment of DNA methylation has become a critical factor for the identification, development and application of methylation based biomarkers. Here we describe a systematic comparison of a quantitative high-resolution mass spectrometry-based approach (MassARRAY), pyrosequencing and the broadly used methylation-specific PCR (MSP) technique analyzing clinically relevant epigenetically silenced genes in acute myeloid leukemia (AML). By MassARRAY and pyrosequencing, we identified significant DNA methylation differences at the ID4 gene promoter and in the 5′ region of members of the SFRP gene family in 62 AML patients compared with healthy controls. We found a good correlation between data obtained by MassARRAY and pyrosequencing (correlation coefficient R2 = 0.88). MSP-based assessment of the identical samples showed less pronounced differences between AML patients and controls. By direct comparison of MSP-derived and MassARRAY-based methylation data as well as pyrosequencing, we could determine overestimation of DNA methylation data by MSP. We found sequence-context dependent highly variable cut-off values of quantitative DNA methylation values serving as discriminator for the two MSP methylation categories. Moreover, good agreements between quantitative methods and MSP could not be achieved for all investigated loci. Significant correlation of the quantitative assessment but not of MSP-derived methylation data with clinically important characteristics in our patient cohort demonstrated clinical relevance of quantitative DNA methylation assessment. Taken together, while MSP is still the most commonly applied technique for DNA methylation assessment, our data highlight advantages of quantitative approaches for precise characterization and reliable biomarker use of aberrant DNA methylation in primary patient samples, particularly.
Introduction
Aberrant epigenetic gene regulation plays a pivotal role in initiation and progression of many different cancer types. Epigenetic levels of transcriptional control comprise a variety of different histone modifications and the addition of a methyl group to the base cytosine. Aberrant DNA methylation, associated with dysregulated gene expression, is a hallmark in virtually all malignant diseases.Citation1 Thus, the assessment of physiological and pathological states of DNA methylation has become the focus of many studies in translational research. Examples include DNA methylation based biomarkers, such as MGMTCitation2,Citation3 or GSTP1Citation4 in glioblastoma or prostate cancer, respectively. These biomarkers have already been introduced into routine clinical diagnostic procedures and contribute to clinical decision making. However, so far there is no clear consensus about the characteristics and requirements of different methodologies for DNA methylation assessment when utilized as biomarker. Currently, several approaches are used for detecting the DNA methylation status at distinct candidate regions. Most of these approaches assess DNA methylation in a qualitative rather than quantitative manner.
Methylation-specific PCR (MSP) is the most commonly used technique for the detection of aberrant DNA methylation in clinical settings, particularly, and has considerably contributed to establishing DNA methylation as a clinically applicable biomarker.Citation5 In a MSP assay, two primer sets containing several CpG dinucleotides (usually 3 or more) are designed for a bisulfite converted target sequence in a methylated and an unmethylated form. MSP is known for very high sensitivity, especially when nested PCR approaches are used.Citation6 Consequently, critical disadvantages of this method are false-positive results and variability of results due to assay conditions (e.g., primer design, annealing temperature, cycle number). In recent years, various MSP-derivatives have been developed including quantitative approaches like MethyLight,Citation7 MS-FLAGCitation8 and others (for a review see ref. Citation9). Semi-quantitative analysis of DNA methylation can be achieved using bisulfite DNA sequencing.Citation10 However, feasibility in routine settings for the assessment of methylation-based biomarkers is restricted due to labor and cost intensity of this method. At present, two major techniques for quantitative DNA methylation analysis of sequence stretches with multiple CpG dinucleotides have emerged. Direct sequencing using the pyrosequencing technique is an accurate and reliably approach for analysis of shorter DNA stretches (usually < 150 bp).Citation11 Pyrosequencing has single nucleotide resolution, high throughput capacity and is highly reproducible. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MassARRAY) provides another new flexible tool for quantitative DNA methylation assessment.Citation12 The biochemical strategy of the MassARRAY technology involves amplification of target regions by PCR after sodium-bisulfite modification of genomic DNA, fragmentation by base-specific cleavage and subsequent analysis of the cleavage products by MALDI-TOF mass spectrometry. Cleavage product signal shifts represent methylation events at single CpGs or small groups of CpGs (CpG units), and signal intensity is correlated with the degree of DNA methylation. Major advantages of this technology are high sensitivity and reproducibility of quantitative measurements, high resolution of target regions and high-throughput capability.
In the present study, we focus on the systematic comparison of MSP-derived methylation data with quantitative high-resolution analysis by pyrosequencing and MassARRAY in acute myeloid leukemia (AML). A previous genome-wide screen for aberrant DNA methylation in AML patients identified the inhibitor of DNA binding 4 (ID4) gene, a member of the helix-loop-helix (HLH) transcription factor family, to be aberrantly methylated in approximately 50% of examined AML samples.Citation13 ID4 has been demonstrated to harbor tumor suppressor activity in various tissues and to be epigenetically silenced in several tumor entities, e.g., colorectal,Citation14 prostate,Citation15,Citation16 breastCitation17 and gastric cancer.Citation18 Furthermore, epigenetic silencing of ID4 has recently been shown to be a pathogenically relevant event also in chronic lymphocytic leukemia (CLL) where quantitative DNA methylation analysis revealed statistically significant correlations between prognostic parameters and DNA methylation level.Citation19
Results
Quantitative DNA methylation analysis of the ID4 promoter
We quantitatively examined the methylation status of ID4 in 62 AML patient samples at diagnosis and 21 samples from healthy donors using the MassARRAY technique. A 401 bp sized amplicon situated in the CpG island covering the transcriptional start site (TSS) and parts of the previously identified core promoter region was investigated ().Citation13 The identical region has recently been detected to be differentially methylated and to harbor prognostic significance in CLL patient samples.Citation19 We observed a wide-spread distribution of markedly elevated DNA methylation levels with a median of 30.5% ranging from 3% to 94% average amplicon methylation in the AML specimens. CpG unit-wise DNA methylation assessment revealed not only substantial differences between different samples but also across the different CpG dinucleotides/CpG units within the analyzed amplicon. Among the AML samples, DNA methylation differences were more pronounced in the 3′ portion (downstream of the TSS) of the investigated amplicon (). In contrast, DNA methylation in various control samples of healthy donors was significantly lower with a median of 7.4% (range 4.4% to 32%) average amplicon methylation (p < 0.0001, ). We could not observe statistically significant differences between different types of healthy controls, namely bone marrow of healthy donors (BM), G-CSF stimulated CD34+ hematopoietic progenitors (CD34+) and cells from buffy coat of the peripheral blood (PB).
Figure 1. Detailed DNA methylation analysis of the ID4 gene 5′ regulatory region. (A) Schematic representation of the ID4 5′ regulatory region including the adjacent CpG island (CGI). The amplicon analyzed by the MassARRAY (MA) assay and by bisulfite sequencing (BS) in AML samples (green) and healthy controls (blue) is depicted as black bar including the position relative to the transcriptional start site (TSS). The regions analyzed by pyrosequencing and methylation-specific PCR are labeled Pyro and MSP (U for reaction with primers specific for unmethylated DNA, M for methylated), respectively. Quantitative MassARRAY results are displayed as heatmap, columns represent single CpG units, rows represent samples. Bright green encodes for low methylation levels, dark blue for high methylation levels. (B) Distribution of average amplicon methylation values by MassARRAY (mC) in AML samples (AML), pooled healthy controls (Ctrl) and separated healthy controls (BM, whole bone marrow; CD34+, G-CSF mobilized CD34 positive hematopoietic progenitor cells from peripheral blood; PB, buffy coat from peripheral blood). Significance was assessed by two-sided non-parametric Wilcoxon Mann-Whitney test. (C) Scatterplot displaying the correlation for mean methylation levels of CpG dinucleotides asessed by both MassARRAY and pyrosequencing. The correlation is calculated as Pearson's R2. (D) Gel electrophoresis of a representative MSP analysis at the ID4 gene 5′ region in AML samples (green) and healthy controls (blue) demonstrating the different MSP-based methylation categories. The location of the amplicon is indicated in . U and M represent the reaction for unmethylated and methylated DNA, respectively. Peripheral blood mononuclear cells (PB), in vitro methylated DNA (IVD) and water (H2O) served as controls. Sample source (BM, PB) and samples IDs are given above the horizontal bars together with the determined methylation category in bold (U, unmethylated; W, weakly methylated; M, methylated). (E) Pie chart showing distribution of MSP-derived DNA methylation categories in healthy controls (n = 18) and AML samples (n = 62).
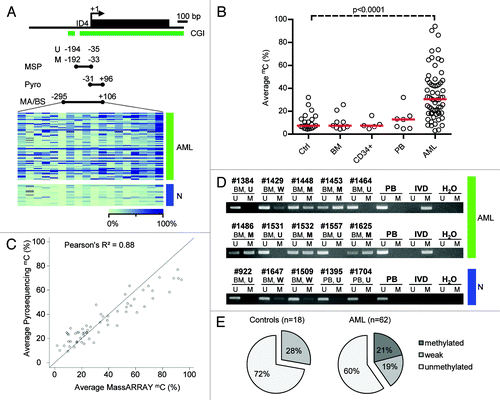
Currently there are mainly two state-of-the-art techniques for quantitative DNA methylation assessment, MassARRAY technology and pyrosequencing. We performed a direct comparison of nine CpG dinucleotides within the described region addressable by both techniques on the identical AML samples (but different batches of bisulfite conversion). Directly comparing identical CpG nucleotides, we found a high correlation (Pearson’s R2 = 0.88) between both methods (and S1). Investigating concordance using Bland-Altman plots, we observed a tendency for higher values below 20–40% methylation for pyrosequencing whereas above 40%, MassARRAY-derived methylation data were slightly elevated (Fig. S2). In addition, we compared both quantitative techniques, MassARRAY and pyrosequencing, to conventional bisulfite sequencing which is still considered as the gold-standard for DNA methylation analyses. We selected five almost unmethylated and five highly methylated samples according to MassARRAY measurements as well as three healthy control samples. Bisulfite sequencing recapitulated the quantitative findings (Fig. S3A–E). For average methylation of the identical CpG dinucleotides, MassARRAY and bisulfite sequencing-derived methylation data were highly correlated (Pearson’s R2 = 0.98). For single CpG units, the correlation ranged from 0.44 to 0.96. A similar good correlation was observed when comparing pyrosequencing and bisulfite sequencing (average methylation: R2 = 0.93, identical single CpG dinucleotides: R2 range 0.48–0.91).
ID4 methylation status in primary AML samples assessed by MSP
In order to compare the MSP technique with quantitative DNA methylation assessment and to explore the discrimination of unmethylated vs. methylated samples, we performed MSP on the identical AML patient set including 18 out of the 21 healthy donors. MSP primers were located inside the region covered by MassARRAY-based methylation analysis flanking the ID4 TSS (). MSP results were divided into three categories: unmethylated (U) in case of presence of a band in the U reaction and absence in the M reaction, methylated (M) in case of a strong band in the M reaction, and low to intermediate methylation (W) if a weak band was reproducibly visible in the M reaction (). The frequency of ID4 methylation among the AML patients as assessed by MSP was 40.3% (25/62 cases), however, 12 out of the 25 (19.3%) methylated cases showed a weak signal for presence of methylation in the M reaction. In contrast, MSP indicated presence of DNA methylation for 27.8% (5/18) of the healthy control samples as demonstrated by weak bands in the M reaction ().
Comparison of MSP-derived DNA methylation data with quantitative approaches
We further aimed at identifying the relation between DNA methylation categories determined by MSP and continuous methylation data by the MassARRAY technique and pyrosequencing. We investigated MassARRAY-derived quantitative DNA methylation levels for all assessable CpG dinucleotides located in the MSP primer sequences (ID4 U+M primers forward: CpG 7, 8 and 9; ID4 U+M primers reverse: CpG 32) and plotted quantitative DNA methylation levels according to the methylation category of the samples for each CpG separately and combined for all CpGs (). Owing to technical reasons, for pyrosequencing, the investigated region did not overlap with the MSP primers but was directly adjacent to the 3′ MSP primer. Therefore, pronounced regional methylation differences could impact on a direct technical comparison between those two methods. For samples evaluated as unmethylated by MSP, the median DNA methylation level assessed by MassARRAY and pyrosequencing was 17% (range 2–37%) and 28% (range 12–66%), for weakly methylated samples 31% (range 8–50%) and 44% (range 10–65%) and for methylated samples 49% (37–91%) and 66.8% (39–89%), respectively (, S4A and B).
Figure 2. Direct comparison of quantitative MassARRAY and MSP-based methylation data at the ID4 5′ region. (A) Distribution of MassARRAY-derived DNA methylation values (mC) for the MSP categories U (unmethylated), W (weakly methylated) and M (methylated) for single CpG units located inside the MSP primer sequences. The MSP forward primer harbors 3 CpG units (identical with MassARRAY CpGs 7–10), for the MSP reverse primer one CpG unit (CpG 32) can be addressed. (B) Distribution of MassARRAY derived DNA methylation values for the MSP categories U, W and M. The CpG units located in the MSP primer sequence are summarized as mean value. All three MSP groups are tested for difference of location using the non-parametric Kruskal-Wallis test. (C) Determination of a MassARRAY based cut-off to classify samples as methylated generating identical proportions of methylated samples for both methods. The Y-axis describes the percentage of samples defined as being methylated. The black curve displays MassARRAY-derived methylation data, the red line represents MSP-based methylation data leading to a cut-off of 42%. (D) Agreement between both methods (inter-rater reliability) by Cohen’s kappa including confidence interval depending on quantitative MassARRAY data.
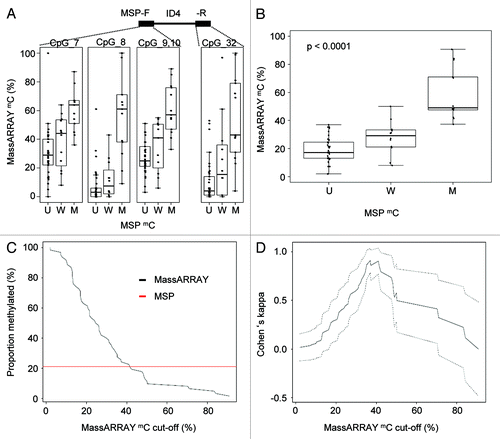
To test which ranges of DNA methylation are covered by qualitative MSP-derived data and to characterize the agreement between the continuous MassARRAY data set with MSP data, we created an overlay of both data sets. As applied in most MSP studies, we treated MSP methylation data as binary data set. This was supported by the similarity between the distributions for the unmethylated (U) and weakly (W) methylated classified samples. Thus, we combined these two categories and compared them to the methylated group (M). This might be particular reasonable considering the risk of overestimating MSP-derived methylation results and therefore of false positive data by excessive sensitivity of MSP as demonstrated in . Both methods gave identical proportions of methylated samples when MassARRAY-derived data above 42% was classified as methylated (). The maximal agreement with a Cohen’s Kappa of 0.91 was reached when MassARRAY methylation levels above 37% were classified as methylated (). The highest prediction accuracy [defined as (true positives + true negatives) / [positives+negatives)] of 0.97 was reached for the cut-off 42% (Fig. S5A and B). For pyrosequencing-derived methylation data, a maximal agreement (Cohen’s Kappa = 0.68) was reached for a cut-off of 59% DNA methylation (Fig. S4A and B).
Classification of MSP-based DNA methylation assessment is locus-dependent
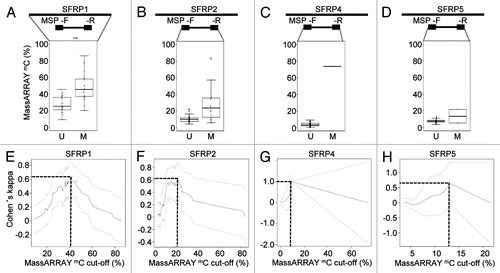
In order to test how the correlation between MSP and quantitative DNA methylation data might change in different regional sequence contexts and in order to explore differences of the agreement and cut-offs defined by both methods,z we extended our analysis to several different loci. We analyzed the methylation status of SFRP1, 2, 4 and 5 using the identical AML patient sample set. Secreted frizzled-related proteins (SFRPs) act as modulators of WNT signaling and have previously been demonstrated to be epigenetically silenced in leukemia.Citation20 All investigated regions had a high CG density and met the criteria of CpG islands.Citation21 We could show that SFRP1 and SFRP2 were significantly methylated in the AML sample set [median 36% (range 9–86%) and 10% (range 3–76%), respectively] compared with the healthy controls [median 15% (range 11–22%) and 7% (range 3–14%), respectively] (Fig. S6). SFRP4 and SFRP5, however, did not show significantly elevated DNA methylation levels neither in the AML patients nor the control cohort. When separating AML samples according to their MSP-based methylation category, samples evaluated as being unmethylated for SFRP1, 2, 4 and 5, showed marked heterogeneity of the median DNA methylation levels (25, 10, 3 and 8% respectively, and S7). Samples detected as methylated by MSP exhibited DNA methylation levels of 45, 24, 73 and 13% for the same regions. However, for SFRP4 and SFRP5, only one and two AML samples, respectively, were judged as methylated by MSP. We could not detect a universally applicable cut-off comparing both methods. Maximal agreements between MSP and MassARRAY according to Cohen’s Kappa analysis were highly variable and strongly dependent on the range of MassARRAY measurements. DNA methylation levels defining optimal agreement with MSP ranged from 9% to 42% (). Most importantly, a good agreement between both methods could not always be achieved as demonstrated by the methylation data of SFRP2 ().
Figure 3. Quantitative DNA methylation analysis of SFRP family members. (A-D) Distribution of MassARRAY-derived DNA methylation values (mC) for the MSP categories U (unmethylated) and M (methylated) for SFRP1 (A), 2 (B), 4 (C) and 5 (D). Distributions of samples categorized as either U or M by MSP were assessed for the average of all CpG units located in MSP primer sequences (indicated as mean F+R). For SFRP1, MSP primers are located in direct vicinity outside of the MassARRAY amplicon, therefore, the mean of the entire MassARRAY amplicon was used for comparison. (E-H) Agreement (inter-rater reliability) between both methods by Cohen’s kappa including confidence interval depending on quantitative MassARRAY data (mC) of all CpG units located in MSP primer sequences (indicated as mean F+R in A-D).
Quantitative DNA methylation assessment is required to reveal correlations of ID4 methylation and AML patient characteristics
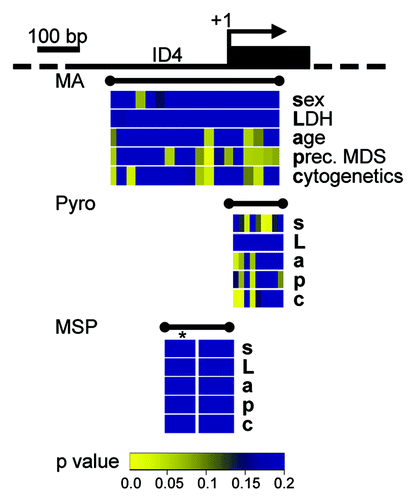
We further tested the ability of high resolution, quantitative DNA methylation assessment and MSP-derived methylation results of the ID4 5′ gene region to reflect correlations with clinical endpoints. Associations between ID4 methylation measurements by MassARRAY, pyrosequencing, and MSP with clinical parameters and known prognostic factors (sex, LDH, age, preceding MDS and cytogenetic risk group) were assessed with univariate non-parametric tests. We could demonstrate that quantitative DNA methylation levels at identical regions of the investigated amplicon assessed by both MassARRAY and pyrosequencing showed a significant correlation with phenotypic features (). A strong correlation of lower DNA methylation levels in AML patients and the presence of a preceding MDS and adverse cytogenetic risk could be detected for CpG dinucleotides situated directly 3′ of the transcriptional start site. Less pronounced correlation of single CpG units could be observed for age and gender. In contrast, MSP-derived methylation data from the same samples did not reveal significant correlations for any of the tested clinical parameters regardless of a potential MSP methylation category ‘weak.’
Figure 4. Quantitative DNA methylation data of the ID4 5′ region reveal correlations with AML disease characteristics. Graphical display of the ID4 gene 5′ region and all raw non-parametric p-values for single the factors sex (s), LDH (L), age (a), presence of a preceding MDS (p) and cytogenetic risk group (c) as derived from univariate testing. Correlations were performed on single CpG units of MassARRAY-derived methylation data as well as for single CpG methylation values by pyrosequencing (Pyro). P-values are displayed in spatial relation to the transcriptional start site (+1). For correlations with MSP-derived DNA methylation status, MSP data were treated either as binary variable (U/M) or using a separation in three methylation states (U/W/M, indicated by *). Color-coding displays low raw p values (significant low p values are depicted in yellow colors; higher p values are represented in a green-blue transition). All p values > 0.2 are displayed in blue.
Discussion
In the current study, we present a thorough comparison of DNA methylation assessment by MSP and quantitative high-resolution techniques, namely MassARRAY and pyrosequencing. MSP is broadly used and preferentially applied in clinical settings, where it supports prognostic stratification and therapeutic decision making (e.g., MGMT in gliblastomaCitation2,Citation3). Therefore we sought to carefully evaluate MSP-derived methylation data using the potential tumor suppressor ID4 as a biologically relevant example in AML and to explore the quantitative DNA methylation levels that are represented by “digitial” MSP categories.
By quantitative means, we demonstrated wide-spread elevated DNA methylation at the ID4 promoter region in AML patients compared with unmethylated or lowly methylated healthy control samples. Elevated DNA methylation levels at the ID4 locus are of considerable interest in the context of AML pathogenesis, first since this gene has recently been characterized as epigenetically silenced tumor suppressor in CLL and second, since its silencing contributes to leukemic progression.Citation19 MassARRAY-derived DNA methylation measurements were highly correlated with pyrosequencing-based data demonstrating that both currently available quantitative state-of-the-art techniques generate concordant results. However, pyrosequencing revealed slightly higher values in methylation levels below 20–40% and lower values above 20–30% compared with MassARRAY. This bias seems to be dependent on the specific amplicon, however, the detailed dynamics of this bias remain unclear. Bisulfite sequencing of identical CpG dinucleotides showed high correlations with both quantitative techniques and validated the obtained results. By MSP, 40% of the AML specimens and 28% of the healthy controls were assigned to be methylated. Thus, MSP data reflected DNA methylation heterogeneity among the group of AML patients but did not reveal similar pronounced differences between AML samples and healthy controls as observed by the quantitative techniques. Particularly, the high proportion of methylation positive samples by MSP among the healthy controls was striking and pointed toward an overestimation of DNA methylation since 84% of healthy controls showed DNA methylation levels below 20% by quantitative measurement.
To further elucidate the direct correlation between MSP-derived methylation categories and quantitative DNA methylation levels, we performed a systematic comparison and searched for potential MSP category thresholds. When categorizing DNA methylation levels by MSP into U (unmethylated), W (weakly methylated) and M (methylated), we detected broad and overlapping ranges of quantitative DNA methylation levels between these categories demonstrating variability and potential for misclassification of MSP-based results. Classification of samples as being weakly methylated and/or methylated started from a median of 8% DNA methylation at the MSP primer sequences. This, however, might explain the high number of samples among the healthy controls being classified as methylated in the MSP results which was not present to a similar degree in the quantitative analysis. The addition of an intermediate methylation category “weak” to the MSP categories did not add considerable information since this category was virtually overlapping with the samples designated as being unmethylated. Similar overlap of DNA methylation levels between MSP categories could be observed for four additional loci (SFRP1, -2, -4 and -5). In conclusion, MSP evaluates a significant proportion of rather lowly methylated samples as being methylated indicating a very high sensitivity even for low levels of DNA methylation. This, on the other hand, might lead to overestimation of DNA methylation. It is also noteworthy that DNA methylation level thresholds between the MSP categories were highly variable and ranged between 10% and 40%. For SFRP2 a clear cut-off could not even be determined. This, however, might indicate that MSP-based classification of DNA methylation could be highly sequence context dependent. Indeed, different MSP amplicons/primers can generate different results and thereby reflect the true methylation state of a given genomic region more or less precisely. Thus, the application of MSP-based DNA methylation changes as a biomarker requires a well-established and tightly controlled assay setup.
Lastly, we demonstrate that quantitative high-resolution DNA methylation assessment can enable identification of correlations with disease phenotype characteristics. Increasing number of reports demonstrate that even single CpG dinucleotides can function as strong prognostic or even predictive indicators which requires detection methods with highest resolution. Here, we show that DNA methylation of several CpG dinucleotides in close vicinity to the ID4 transcriptional start site assessed by both quantitative techniques correlates with cytogenetic risk status. Lower DNA methylation levels in this region are preferentially found in patients with secondary AML from preceding MDS. This observation is interesting since it might point toward a role of epigenetic alteration at the ID4 locus in the process of leukemic progression. A similar context for ID4 methylation was reported by a previous report describing increased ID4 methylation assessed by MSP in MDS as a marker for higher risk of leukemic transformation.Citation22
In conclusion, our results identify distinct properties of the quantitative techniques and of MSP which implies specific fields of application for the respective methods. We propose a strong rationale for high-resolution quantitative DNA methylation measurements as gold-standard for de novo DNA methylation assessment. Both quantitative state-of-the-art techniques, MassARRAY and pyrosequencing, generate highly similar results. Particularly in translational research, sample purity and contaminating cell populations with (tissue-) specific methylation patterns might lead to false positive detection of DNA methylation. Here, MSP can cause overestimation of DNA methylation frequencies. Furthermore, continuous DNA methylation data can reveal associations (e. g. phenotypical disease features) that are not detectable by categorized measurements, since dichotomizing usually leads to loss of power.Citation23 These fundamental assay characteristics, however, represent key prerequisites for further refined successful biomarker development. The quantitative nature will make it easier to transfer technologies to different labs and to create clinical standards. Quantitative techniques have entered the DNA methylation field and are becoming available for translational research questions and even for routine application in clinical settings. On the other hand, specific MSP assays for distinct genomic regions are tested and well established and generate robust and reliable results. MSP will remain of considerable importance where exquisite assay sensitivity is required or only low DNA amounts are available such as detection of hypermethylated sequences in biological fluids, serums and biopsies.
Material and methods
Patients and samples
A total of 62 DNA samples derived from 19 peripheral blood (PB) and 43 bone marrow (BM) specimens from adult AML patients were provided by the University hospital of Aachen with patient informed consent and institutional review board approval in accordance with the Declaration of Helsinki. Myeloblasts were enriched by separating mononuclear cells from BM and PB using Ficoll density gradient. Genomic DNA was isolated using the QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer’s instructions. The median follow-up time was 10.3 mo. Detailed clinical information is provided in . Additionally, buffy coat from the PB of seven healthy donors, five stem cell harvests obtained by leukapheresis from patients with solid tumors or non Hodgkin’s lymphoma without evidence for BM involvement as well as nine BM samples from patients without evidence for malignant hematological disease were analyzed.
Table 1. AML patient characteristics
Methylation-specific polymerase chain reaction
Methylation-specific polymerase chain reaction (MSP) analysis was used for qualitative analysis of methylation as previously described.Citation24 Briefly, 1 µg of DNA was sodium bisulfite-modified using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturer’s instructions. Polymerase chain reaction (PCR) amplification of the bisulfite-converted DNA was performed in two separate reactions with primer pairs specific for either the unmethylated (U) or the methylated (M) sequence. MSP primer sequences are given in Table S1. Reactions were hot-started at 95°C for 5 min and maintained at 80°C before adding 0.625 U of Taq Polymerase (Sigma). Temperature conditions for PCR were as follows: 35 cycles of 58C for 30 sec, primer-specific annealing temperature for 30 sec and 72°C for 30 sec. DNA from PB was treated in vitro with M.SssI methyltransferase (New England Biolabs) to generate a fully methylated control. PCR products were separated on 2.5% agarose gels and visualized by ethidium bromide staining.
Quantitative DNA methylation by MassARRAY technology
Quantitative DNA methylation analysis was performed using MALDI-TOF-MS–based approach (referred to as MassARRAY in the manuscript) by Sequenom as described previously.Citation12 Target regions for DNA methylation analysis were designed to yield maximum information for single CpG dinucleotides by in silico processing using custom R-based scripts. Primer sequences are given in Table S1. In brief, the target gene regions were amplified by PCR after sodium-bisulfite modification of genomic DNA. Subsequently deoxynucleotides in the PCR reaction were inactivated by dephosphorylation using shrimp alkaline phosphatase (SAP). By tagging the reverse PCR primer with the T7 recognition sequence, a single-stranded RNA copy of the template was generated by in vitro transcription. After base specific (U-specific) cleavage by RNase A, the cleavage products were then analyzed using MALDI-TOF mass spectrometry. Cleavage product signals with a 16 Da shift (or a multiple thereof) are representative for methylation events, and signal intensity is correlated with the degree of DNA methylation.
Pyrosequencing
Pyrosequencing was conducted with bisulfite modified DNA to analyze and quantify the methylation status of the DNA. With the first PCR primer pair, the methylated and the unmethylated genomic DNA of the ID4 gene were amplified (primer sequences are given in Table S1). A single strand linear DNA was prepared from the PCR product with the PyroMark TM Vacuum Prep Workstation (Qiagen) that permits the isolation of the DNA strand with the biotinylated primer. The sequencing PCR was performed with a gene specific sequencing primer on a PyroMark Q96 ID System (Qiagen). During synthesis of the cDNA strand, a luciferase-catalyzed lightpulse was generated upon incorporation of the new nucleotides. This photometrically measured light pulse is in proportion to the number of incorporated nucleotides and thereby quantitatively displays the methylation of the investigated CpG dinucleotides. For analysis, the software PyroMark Q CpG software (Qiagen) was used. For primer design, PSQ Assay Design, Version 1.0 (Qiagen) was used.
Bisulfite sequencing
Five patient samples with high and low ID4 methylation according to MassARRAY-derived data, respectively, and three healthy controls were selected for bisulfite sequencing. The ID4 amplicon analyzed by MassARRAY containing 50 CpG dinucleotides was cloned using the TOPO TA Cloning Kit (Invitrogen) following the manufacturer’s instructions. In brief, the ID4 region of interest was PCR amplified with the identical primer set as used for MassARRAY analyses. The amplified DNA was then purified and ligated into the TOPO pCR2.1 vector. After precipitation, the ligation product was transformed into electrocompetent E. coli cells. Bacteria were grown under ampicillin selection on LB plates. Per sample, 12 colonies were picked for plasmid purification and subsequent Sanger sequencing. Sequences of ten clones per sample were analyzed using the BISMA software suite.Citation25
Statistical methods
Samples with a proportion of missing MassARRAY- and pyrosequencing-derived methylation data exceeding 20% were excluded from further analysis. Remaining missing measurements were imputed. MSP was either analyzed as yes/no variable with ’weak’ being considered no methylation, or with ’weak’ as intermediate category. The association between MSP and quantitative measurements was tested with the non-parametric Wilcoxon Mann-Whitney and Kruskall-Wallis test. Binary MSP and MassARRAY/pyrosequencing measurements were compared for inter-rater reliability and agreement with Cohen’s kappa coefficient including pointwise 95% confidence intervals.Citation26 As it is not possible to directly determine the agreement between binary and continuous data, all possible cut-offs of MassARRAY/pyrosequencing data were computed in order to compare two types of dichotomous methylation results. Thus, agreement was calculated as a function of the MassARRAY/pyrosequencing cut-off. In addition, receiver operating characteristic (ROC) and prediction accuracy analyses were performed to compute the best possible separation of MSP measurements with regard to a MassARRAY cut-off. These calculations were done for each CpG unit targeted by the MSP primer separately, the average of them as well as the average of the whole amplicon. Agreement between MassARRAY and pyrosequencing was assessed with Bland-Altman plots including 95% limits of agreement. Association between methylation and clinical covariates was assessed univariately for each CpG separately with non-parametric methods. We used Spearman’s correlation coefficient for continuous response variables (age, LDH), Wilcoxon-Mann-Whitney test for binary traits (preceding MDS, sex) and the Jonckheere-Terpstra test for ordinal responses (cytogenetic risk group). Association between MSP and clinical covariates was assessed with Fisher’s exact test for categorical traits and Wilcoxon-Mann-Whitney test for continuous parameters. Since p values were primarily used to describe and compare strength of association between both methylation methods, no adjustment for multiplicity is applied. P values below 0.05 were considered statistically significant. All statistical analyses were performed using the R statistical environment version 2.10.0 including add-on package rms and Hmisc.Citation27
| Abbreviations: | ||
| AML | = | acute myeloid leukemia |
| BM | = | bone marrow |
| BS | = | bisulfite sequencing |
| bp | = | base pairs |
| CLL | = | chronic lymphocytic leukemia |
| CpG | = | CpG dinucleotide |
| Da | = | Dalton |
| FAB | = | French-American-British (classification) |
| G-CSF | = | granulocyte colony stimulating factor |
| HLH | = | helix-loop-helix |
| LB | = | lysogeny broth (medium) |
| LDH | = | lactate dehydrogenase |
| MALDI-TOF | = | matrix-assisted laser desorption ionization time-of-flight |
| MDS | = | myelodysplastic syndrome |
| MS-FLAG | = | methylation-specific fluorescent amplicon generation |
| MSP | = | methylation-specific polymerase chain reaction |
| PB | = | peripheral blood |
| PCR | = | polymerase chain reaction |
| ROC | = | receiver operating characteristic |
| SAP | = | shrimp alkaline phosphatase |
| TSS | = | transcriptional start site |
| U | = | M, W, MSP-derived methylation categories unmethylated, methylated, weakly methylated |
| WBC | = | white blood cells |
Additional material
Download Zip (566.8 KB)Acknowledgments
We thank Prof. Dr. Tim H. Brümmendorf for generous support with patient samples and cinical data as well as constructive discussions. We are grateful to Oliver Mücke, Marion Bähr and Olga Bogatyrova (German Cancer Research Center, Heidelberg, Germany) for excellent technical support with MassARRAY-based methylation analyses and bisulfite sequencing. The study was supported by a fellowship from the German Research Council (DFG) to RC (CL 427/2–1). CP is supported by NIH grant CA101956. EJ received grant support from the German Cancer Aid.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 92; http://dx.doi.org/10.1016/j.cell.2007.01.029; PMID: 17320506
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350 - 4; http://dx.doi.org/10.1056/NEJM200011093431901; PMID: 11070098
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352:997 - 1003; http://dx.doi.org/10.1056/NEJMoa043331; PMID: 15758010
- Richiardi L, Fiano V, Vizzini L, De Marco L, Delsedime L, Akre O, et al. Promoter methylation in APC, RUNX3, and GSTP1 and mortality in prostate cancer patients. J Clin Oncol 2009; 27:3161 - 8; http://dx.doi.org/10.1200/JCO.2008.18.2485; PMID: 19470943
- Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93:9821 - 6; http://dx.doi.org/10.1073/pnas.93.18.9821; PMID: 8790415
- Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res 2000; 60:5954 - 8; PMID: 11085511
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res 2000; 28:E32; http://dx.doi.org/10.1093/nar/28.8.e32; PMID: 10734209
- Bonanno C, Shehi E, Adlerstein D, Makrigiorgos GM. MS-FLAG, a novel real-time signal generation method for methylation-specific PCR. Clin Chem 2007; 53:2119 - 27; http://dx.doi.org/10.1373/clinchem.2007.094011; PMID: 17962364
- Kristensen LS, Hansen LL. PCR-based methods for detecting single-locus DNA methylation biomarkers in cancer diagnostics, prognostics, and response to treatment. Clin Chem 2009; 55:1471 - 83; http://dx.doi.org/10.1373/clinchem.2008.121962; PMID: 19520761
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 1994; 22:2990 - 7; http://dx.doi.org/10.1093/nar/22.15.2990; PMID: 8065911
- Tost J, Dunker J, Gut IG. Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. Biotechniques 2003; 35:152 - 6; PMID: 12866415
- Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A 2005; 102:15785 - 90; http://dx.doi.org/10.1073/pnas.0507816102; PMID: 16243968
- Yu L, Liu C, Vandeusen J, Becknell B, Dai Z, Wu YZ, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet 2005; 37:265 - 74; http://dx.doi.org/10.1038/ng1521; PMID: 15723065
- Gómez Del Pulgar T, Valdés-Mora F, Bandrés E, Pérez-Palacios R, Espina C, Cejas P, et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int J Oncol 2008; 33:185 - 93; PMID: 18575765
- Carey JP, Asirvatham AJ, Galm O, Ghogomu TA, Chaudhary J. Inhibitor of differentiation 4 (Id4) is a potential tumor suppressor in prostate cancer. BMC Cancer 2009; 9:173; http://dx.doi.org/10.1186/1471-2407-9-173; PMID: 19500415
- Asirvatham AJ, Schmidt MA, Chaudhary J. Non-redundant inhibitor of differentiation (Id) gene expression and function in human prostate epithelial cells. Prostate 2006; 66:921 - 35; http://dx.doi.org/10.1002/pros.20366; PMID: 16541417
- Noetzel E, Veeck J, Niederacher D, Galm O, Horn F, Hartmann A, et al. Promoter methylation-associated loss of ID4 expression is a marker of tumour recurrence in human breast cancer. BMC Cancer 2008; 8:154; http://dx.doi.org/10.1186/1471-2407-8-154; PMID: 18513385
- Chan AS, Tsui WY, Chen X, Chu KM, Chan TL, Chan AS, et al. Downregulation of ID4 by promoter hypermethylation in gastric adenocarcinoma. Oncogene 2003; 22:6946 - 53; http://dx.doi.org/10.1038/sj.onc.1206799; PMID: 14534543
- Chen SS, Claus R, Lucas DM, Yu L, Qian J, Ruppert AS, et al. Silencing of the inhibitor of DNA binding protein 4 (ID4) contributes to the pathogenesis of mouse and human CLL. Blood 2011; 117:862 - 71; http://dx.doi.org/10.1182/blood-2010-05-284638; PMID: 21098398
- Jost E, Schmid J, Wilop S, Schubert C, Suzuki H, Herman JG, et al. Epigenetic inactivation of secreted Frizzled-related proteins in acute myeloid leukaemia. Br J Haematol 2008; 142:745 - 53; http://dx.doi.org/10.1111/j.1365-2141.2008.07242.x; PMID: 18537968
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196:261 - 82; http://dx.doi.org/10.1016/0022-2836(87)90689-9; PMID: 3656447
- Wang H, Wang XQ, Xu XP, Lin GW. ID4 methylation predicts high risk of leukemic transformation in patients with myelodysplastic syndrome. Leuk Res 2010; 34:598 - 604; http://dx.doi.org/10.1016/j.leukres.2009.09.031; PMID: 19853913
- Altman DG, Royston P. The cost of dichotomising continuous variables. BMJ 2006; 332:1080; http://dx.doi.org/10.1136/bmj.332.7549.1080; PMID: 16675816
- Galm O, Herman JG. Methylation-specific polymerase chain reaction. Methods Mol Med 2005; 113:279 - 91; PMID: 15968111
- Rohde C, Zhang Y, Reinhardt R, Jeltsch A. BISMA--fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics 2010; 11:230; http://dx.doi.org/10.1186/1471-2105-11-230; PMID: 20459626
- Cohen J. A coefficient of agreement for nominal scales.. Educational and Psychological Measurement 1960; 20:37 - 46
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2012.