Abstract
Developmental regulation of gene expression is controlled by distinct epigenetic signatures catalyzed by various epigenetic modifiers. Little is known about the ontogeny and tissue distribution of these epigenetic modifiers. In the present study, we used a novel approach of RNA-sequencing to elucidate hepatic ontogeny and tissue distribution of mRNA expression of 142 epigenetic modifiers, including enzymes involved in DNA methylation/demethylation, histone acetylation/deacetylation, histone methylation/demethylation, histone phosphorylation and chromosome remodeling factors in male C57BL/6 mice. Livers from male C57BL/6 mice were collected at 12 ages from prenatal to adulthood. Many of these epigenetic modifiers were expressed at much higher levels in perinatal livers than adult livers, such as Dnmt1, Dnmt3a, Dnmt3b, Apobec3, Kat1, Ncoa4, Setd8, Ash2l, Dot1l, Cbx1, Cbx3, Cbx5, Cbx6, Ezh2, Suz12, Eed, Suv39h1, Suv420h2, Dek, Hdac1, Hdac2, Hdac7, Kdm2b, Kdm5c, Kdm7, Prmt1–5, Prmt7, Smarca4, Smarcb1, Chd4 and Ino80e. In contrast, hepatic mRNA expression of a few epigenetic modifiers increased during postnatal liver development, such as Smarca2, Kdm1b, Cbx7 and Chd3. In adult mice (60 d of age), most epigenetic modifiers were expressed at moderately (1–3-fold) higher levels in kidney and/or small intestine than liver. In conclusion, this study, for the first time, unveils developmental changes in mRNA abundance of all major known epigenetic modifiers in mouse liver. These data suggest that ontogenic changes in mRNA expression of epigenetic modifiers may play important roles in determining the addition and/or removal of corresponding epigenetic signatures during liver development.
Introduction
Liver, kidney, and intestine are the three major tissues involved in the absorption, distribution, metabolism, and elimination of xenobiotics. Many drug-processing genes undergo marked changes in expression during tissue development and maturation; epigenetic modifications play a key role in developmental regulation of gene expression.Citation1 Currently, the knowledge on epigenetic regulation of drug processing genes is very limited.Citation2 The addition and removal of various epigenetic signatures, namely DNA methylation and histone modifications, are mediated by various epigenetic enzymes and chromosome remodeling factors. Elucidation of developmental changes in expression patterns of these epigenetic modifiers will help to understand the mechanism of epigenetic regulation of ontogenic gene expression. Additionally, tissue-specific gene expression patterns are determined by distinct epigenetic signatures,Citation3 however, it is not clear whether tissue-specific expression of epigenetic enzymes contributes to tissue-specific epigenetic profiles. Therefore, the purpose of the present study was to elucidate ontogenic changes in mRNA expression of various epigenetic modifiers during liver development and the distribution of mRNA expression of these epigenetic modifiers in adult tissues of liver, kidney, and small intestine.
DNA methylation is involved in proper gene regulation, chromosomal stability, and parental imprinting.Citation4,Citation5 Histone proteins in nucleosomes, surrounded by DNA, play a key role in the regulation of gene expression. Several covalent modifications on the N-termini of histone proteins have been discovered, many of which contain distinct regulatory functions.Citation6 Modifications can be divided into those that correlate with activation, and those that correlate with repression. Acetylation, methylation, phosphorylation, and ubiquitination have been implicated in activation, whereas methylation, ubiquitination, sumoylation, deamination, and proline isomerization have been implicated in repression.Citation6
Methylation of histones by methyltransferases can lead to various effects in gene transcription, depending on which lysine is methylated and the level of methylation (mono-, di-, tri-methylation) occur. By analyzing a chromatin landmark and transcription initiation in most promoters in human ES cells, 3 classes of genes with various histone methylations have been identified.Citation7 The majority of actively transcribed genes have nucleosomes modified by di- or tri-methylation of histone H3 at lysine 4 (H3K4me2 or 3) around the transcription start sites (TSS), and histone H3 trimethylation at lysine 36 (H3K36me3) along the gene coding regions after the TSSs. Certain genes do not have H3K4me2/3-modified nucleosomes around their TSS; some of them are enriched with trimethylation of histone H3 at lysine 27 (H3K27me3) along the genes. A recent ChIP-on-chip study demonstrates that H3K27me3 forms broad local enrichments over silent genes and intergenic regions on gene-rich regions on mouse chromosome 17.Citation8 Vast data have confirmed that H3K4me2 and H3K4me3 are related to initiation of gene transcription, H3K36me3 is correlated with elongation of gene transcription, whereas H3K27me3 is associated with suppression of gene transcription.Citation9-Citation15 Methylation and demethylation of H3K4, H3K36, and H3K27 serve as a “multistable-switch” model of epigenetic memory to control gene expression in cellular differentiation and development.Citation16
Dimethylation of histone H3 at lysine 9 (H3K9me2) is widespread in chromatin, frequently associated with DNA methylation, and responsible for stable gene silencing in heterochromatin and reversible gene suppression in euchromatin.Citation17-Citation19 Approximately 46% of the mouse genome is covered by H3K9me2 in adult liver, and large H3K9me2-modified DNA regions are associated with tissue-specific gene silencing in differentiated tissues.Citation20 Additionally, trimethylation of histone H3 at lysine 9 (H3K9me3) is associated with gene silencing in heterochromatin.Citation21
The addition and removal of DNA methylation and histone modifications are catalyzed by various epigenetic modifiers. Results from studies of various knockout mice demonstrate that there is often overlapping in substrates among these epigenetic enzymes. Therefore, to elucidate the mechanism of epigenetic regulation of gene expression, it is important to understand which epigenetic enzymes are expressed in a given tissue during a given developmental stage. Currently, little is known about the ontogenic expression patterns, tissue distribution, and regulatory mechanisms of these key epigenetic enzymes. Conventional mRNA-profiling methods cannot compare the real abundance of transcripts between different genes. In contrast, RNA-sequencing (RNA-seq) provides a “true-quantification” of transcript counts.Citation22 Therefore, the purpose of this study was to use RNA-seq technology to generate comprehensive information on the ontogenic mRNA expression of all major known epigenetic modifiers in liver and the distribution of mRNAs of these genes in adult liver, kidney, and small intestine using mouse as a model, which will provide a foundation for determining the regulatory mechanisms controlling the various transcription patterns of these epigenetic modifier genes.
Results
The total FPKM values of all mRNA transcripts determined by RNA-seq were very similar among livers from different ages of mice (Fig. S1A) or among the three adult mouse tissues of liver, kidney, and small intestine (Fig. S1B), which is consistent with the equal loading of total RNAs from these samples in RNA-seq. The RNA-seq data of ontogenic mRNA expression in livers of C57BL/6 mice were highly quantitative, which had been validated in our previous studies.Citation23,Citation24 The present RNA-seq data of distribution of mRNA expression in liver, kidney, and small intestine of adult male C57BL/6 mice were highly consistent with the known knowledge about tissue-specific/predominant gene expression in these three tissues (Fig. S2), demonstrating the high qualities of tissue samples and the reliability of RNA-seq data.
Ontogeny and tissue distribution of mRNAs of genes involved in DNA methylation and demethylation
DNA methyltransferase 1 (Dnmt1) is a key maintenance DNA methyltransferase that predominantly methylates hemi-methylated CpG dinucleotides, whereas Dnmt3a and Dnmt3b methylate hemi-methylated and unmethylated CpG at the same rate. Dnmt1 mRNA was much more abundant in fetal liver than Dnmt3a/3b, and it sharply decreased during postnatal liver development, resulting in ~5% of the fetal level in adulthood (). Hepatic Dnmt3a and Dnmt3b mRNAs also decreased 60–70% during postnatal development (). Compared with adult liver, adult kidney and intestine had 1–2 fold higher levels of Dnmt1 and Dnmt3a mRNAs, but ~40% less Dnmt3b mRNA (). DNA (cytosine-5)-methyltransferase 3-like (Dnmt3l) mRNA was barely detectable during liver development as well as in adult kidney and intestine (data not shown).
Figure 1. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in DNA methylation and demethylation in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
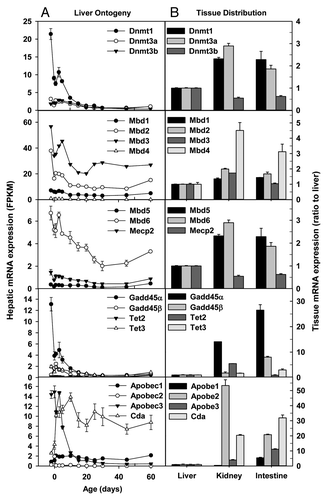
Methyl-CpG binding domain proteins (Mbd) are a highly conserved family of DNA-binding proteins that convert the information represented by methylation patterns into the functional state. Mbd2 and Mbd3 were the most abundant Mbds in fetal and adult livers, followed by Mbd1 and Mbd6, with Mbd4, Mbd5, and Mecp2 expressed at low levels during liver development (). Hepatic Mbd2 and Mbd3 mRNAs were the highest at days before birth, and decreased moderately (~50%) during postnatal development. Mbd1, Mbd4, Mbd6, and Mecp2 mRNAs were also moderately downregulated during postnatal liver development, whereas Mbd5 had no ontogenic changes. Mbds were generally expressed at higher levels in adult kidney and intestine than adult liver ().
The methylation of DNA can be removed by enzymes that catalyze demethylation of DNA. Family members of growth arrest and DNA-damage-inducible protein 45 (Gadd45), ten-11 translocation (Tet), and activation-induced cytidine deaminase/(AID)/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (Apobec) proteins are involved in active and passive DNA demethylation.Citation25,Citation26 Gadd45α mRNA was much higher in fetal livers than adult livers; in contrast, Gadd45β tended to be expressed more during perinatal stage (), and Gddd45γ mRNA remained very low throughout liver development (data not shown). Among Tet family members, only Tet3 had detectable mRNA expression in liver, and hepatic Tet3 mRNA decreased 50–70% during postnatal development (). In adulthood, Gadd45α and Gadd45β mRNAs were expressed at 14–26 and 1–7 fold higher levels, respectively, in kidney and small intestine than liver (). In adulthood, Tet2 and Tet3 mRNAs were expressed the highest in kidney and intestine, respectively. Among Apobec1–3, Apobec3 had the highest expression in fetal liver; its expression sharply decreased during postnatal liver development. In contrast, hepatic mRNA expression of cytidine deaminase (Cda) surged 135% 1 d after birth, and remained relatively constant thereafter (). Compared with adult liver, adult kidney and small intestine had much higher mRNA expression of Apobecs and Cda ().
Ontogeny and tissue distribution of mRNAs of genes involved in histone acetylation
Family members of lysine acetyltransferase (Kat) catalyze the acetylation of histones and non-histone targets. Hepatic mRNA expression of Kat1/Hat1 sharply decreased 89% from pre-natal to adulthood, whereas that of Kat2a/Gcn5 decreased 57% (). In contrast, hepatic Kat2b/Pcaf mRNA tended to increase during postnatal development, whereas Kat3b/p300 () and Kat3a mRNAs (data not shown) remained low throughout liver development. In adulthood, Kat1 mRNAs were 1–1.5 fold higher in kidney and small intestine than liver (). Kat2a and Kat3b mRNAs were moderately higher in kidney and small intestine than liver, whereas Kat2b mRNA was ~50% lower in small intestine than liver ().
Figure 2. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in histone acetylation in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
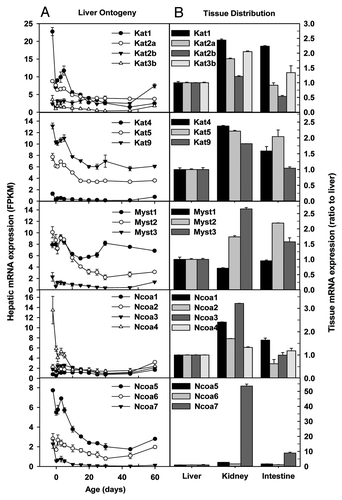
Hepatic Kat4/Taf1 mRNA remained low throughout liver development, whereas Kat5/Tip60 and Kat9/Elp3 mRNAs decreased ~50% from prenatal to adulthood (). In adulthood, Kat4, Kat5, and Kat9 mRNAs in kidney and small intestine were ~50–150% higher than those in liver ().
Histone acetyltransferases of the MYST family are important in stem cells, development, and tumorigenesis.Citation27 Hepatic mRNA expression of Myst1/Kat8 and Myst3/Kat6a fluctuated throughout liver development, whereas that of Myst2/Kat7 gradually decreased (). Myst1 mRNA levels were similar in the 3 adult tissues, whereas Myst2 and Myst3 mRNAs were moderately higher in adult kidney and small intestine than adult liver ().
Family members of the nuclear receptor coactivator (Ncoa) function as coactivators of nuclear receptors and certain other transcription factors. Certain Ncoa, such as Ncoa1/Src1, has intrinsic histone acetyltransferase activity, whereas other Ncoa can recruit histone acetyltransferases or methyltransferases to activate gene expression.Citation28 Hepatic mRNAs of Ncoa1/Src1, Ncoa2/Src2, and Ncoa3/Src3 remained consistently low throughout liver development (). In contrast, Ncoa4/ARA70 mRNA sharply decreased within 10 d post birth, resulting in a very low mRNA expression in adulthood, similar to other Ncoa1–3 family members (). Adult kidney had moderately higher Ncoa1–4 mRNAs than adult liver, whereas adult intestine and liver had comparable amounts of Ncoa1–4 mRNAs ().
Hepatic Ncoa5/Cia mRNA decreased ~50–70% during postnatal development, whereas Ncoa6/Asc2 mRNA fluctuated at low levels during liver development (). Ncoa7/Erap140 is a conserved tissue-specific nuclear receptor coactivator predominantly expressed in brain and kidney.Citation29 Hepatic Ncoa7 mRNA was only expressed significantly in fetal liver (). Consistent with a previous report,Citation29 Ncoa7 mRNA was expressed at much higher levels in adult kidney than adult small intestine and liver ().
Ontogeny and tissue distribution of mRNAs of genes involved in histone deacetylation
There are two protein families with histone deacetylase (HDAC) activity: the classical HDAC family and the recently discovered SIRT family of NAD+-dependent HDACs.Citation30 Members of the classical HDAC family fall into 2 classes, namely class I (HDAC1, 2, 3 and 8) and class II (HDAC4, 5, 6, 7, 9 and 10). Presently, it is thought that HDACs of class I are expressed in most cell types, whereas the expression of class II HDACs is more restricted, suggesting their role in cellular differentiation and developmental processes. In regard to class I Hdacs, hepatic mRNAs of Hdac1, Hdac2, and Hdac3 were 2–3 fold higher in perinatal livers than adult livers, whereas Hdac8 mRNA was consistently low throughout liver development (). The mRNA expression of class IIa Hdac5 fluctuated at moderate levels, whereas that of Hdac7 decreased sharply during postnatal liver development. Hdac4 and Hdac9 mRNAs were barely detectable throughout liver development (). In regard to the Hdac IIb family, Hdac6 mRNA reached the highest right after birth, and gradually decreased to low levels in adulthood, whereas Hdac10 mRNA remained little changed during liver development (). The SIRT family displayed diverse expression patterns during liver development (). Sirt1, Sirt4, Sirt5, and Sirt6 mRNAs were low, with moderate decreases in Sirt1 and Sirt6 but increases in Sirt4 and Sirt5 observed during postnatal development. In contrast, Sirt2 and Sirt7 mRNAs were consistently expressed at moderate levels throughout liver development. Hepatic Sirt3 and Hdac11 mRNAs surged 3-fold during perinatal stage, and fluctuated at moderate levels thereafter ().
Figure 3. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in histone deacetylation in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
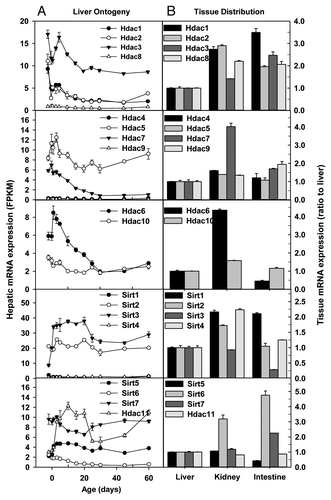
The mRNA expression of Hdac family varied in the 3 adult tissues (). Class I and IIb Hdac mRNAs were expressed at 1–2 fold higher levels in adult kidney and small intestine than adult liver. The class IIb Hdac6 mRNA had the most diverse expression pattern: Hdac6 mRNA in kidneys was 3-fold higher than that in liver, whereas Hdac6 mRNA in small intestine was only ~50% of that in liver (). In adulthood, Sirt mRNAs tended to be slightly higher in kidney and small intestine than liver, except that Sirt3 and Sirt5 mRNAs in small intestines were only 25–50% of those in liver. Hdac11 mRNA was similarly expressed at moderate levels in the 3 adult tissues ().
Ontogeny and tissue distribution of mRNAs of genes involved in histone methylation
Family members of SET domain containing (Setd) are histone/lysine methyltransferases. Hepatic Setd1a and Setd3 mRNAs decreased ~50% after birth, but slightly increased again when reaching adulthood (). In contrast, hepatic Setd2 and Setd4 mRNAs remained consistently low throughout liver development (). In adulthood, Setd1a, 2, 3, and 4 mRNAs were comparable between liver and small intestine, but were 50–100% higher in kidney than liver, with the exception of a ~50% lower Setd4 mRNA in kidney than liver ().
Figure 4. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in histone methylation in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
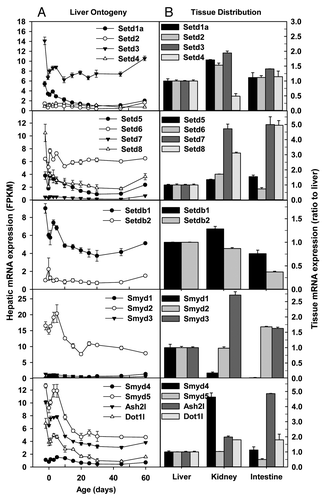
Hepatic mRNA expression of Setd5, Setd6, and Setd7 fluctuated during liver development, whereas hepatic Setd8 mRNA sharply decreased 60–80% after birth and was only 35% of fetal level in adulthood (). In adulthood, Setd5 and Setd6 mRNAs were similar among the 3 tissues, whereas Setd7 and Setd8 mRNAs in kidney and small intestine were 2–4 fold higher than liver ().
Hepatic Setdb1 mRNA decreased ~50% during postnatal development, whereas Setdb2 mRNA remained low throughout liver development (). In adulthood, Setdb1 mRNA expression was similar among the 3 tissues, and Setdb2 mRNA was lowly expressed in all 3 tissues ().
Family members of SET and MYND domain containing (Smyd) are also histone/lysine methyltransferases. Smyd1, Smyd3, and Smyd4 mRNAs were consistently expressed at low levels throughout liver development (). In contrast, Smyd2 and Smyd5 had the highest expression during the perinatal stage, and remained readily detectable in adulthood. Ash2l regulates histone H3K4 trimethylation,Citation31 whereas Dot1l specifically catalyzes H3K79 methylation.Citation32 Hepatic Ash2l and Dot1l mRNAs were expressed highest before birth and decreased to low levels in adulthood (). In adulthood, Smyd1 mRNA was very low, whereas Smyd2 mRNA was comparable, among the 3 tissues. Smyd3 and Smyd4 mRNAs were the highest in kidney, whereas Ash2l mRNA highest in small intestine ().
Euchromatin histone methyltransferase 1 (Ehmt1/Glp) and Ehmt2/G9a form heteromeric complexes and are essential in catalyzing H3K9me2, a key silencing epigenetic signature.Citation33 Glp mRNA was consistently low throughout liver development, whereas G9a mRNA in perinatal liver was twice that in adulthood ().
Figure 5. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in histone methylation and recognition of methylated histones in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
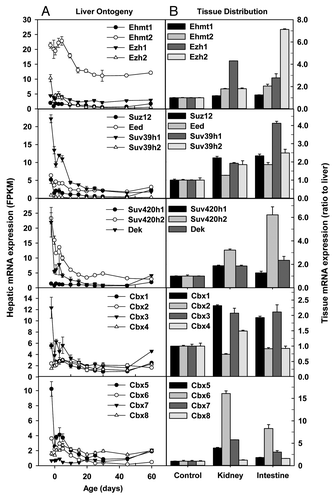
Polycomb group (PcG) proteins are the most well characterized repressors of gene expression. PcG proteins are essential for normal cell proliferation, and have been implicated in the maintenance of stem cells. In Drosophila, PcG proteins form two major complexes, polycomb repressive complex 1 (PRC1) and PRC2. Enhancer of zeste homolog 1 (Ezh1) and Ezh2 catalyze H3K27me3, a key silencing epigenetic signature. Ezh1 mRNA was little changed during liver development, whereas hepatic Ezh2 mRNA was sharply downregulated after birth, resulting in very low expression in adulthood (). EZH2, together with SUZ12 and EED, forms PRC2, which catalyzes H3K27me3.Citation34 Similar to Ezh2, hepatic Suz12 and Eed mRNAs were highest in prenatal liver, downregulated after birth, but slightly rebounded in adulthood (). The 3 adult tissues of liver, kidney, and small intestine had comparable mRNA expression of Ehmt1, Ehmt2, Suz12 and Eed (). In contrast, Ezh1 mRNA was ~3 fold higher in adult kidney than adult liver, whereas adult small intestine had ~6 fold higher Ezh2 mRNA than adult liver ().
Suv39h1 is essential in maintaining H3K9 methylation at pericentric heterochromatin.Citation35 Suv39h2 is another histone H3 methyltransferase that shares 59% identity with Suv39h1 and displays testis-specific expression.Citation36 SUV420H proteins catalyze H4K20 methylation and differ substantially in their localization and activity.Citation37 Dek is a Suppressor of Variegation [Su(var)] that is essential for heterochromatin integrity through interacting directly with Hp1α and markedly enhancing its binding to H3K9me3.Citation38 Hepatic mRNAs of Suv39h1, Suv420h2, and Dek sharply decreased after birth, resulting in low mRNA levels in adulthood (). In contrast, hepatic mRNA expression of Suv39h2 and Suv420h1 was consistently low throughout liver development. Suv420h1 and Dek mRNAs were low in the 3 adult tissues of liver, kidney, and small intestine, whereas Suv420h2 mRNA was ~5 fold higher in small intestine than liver ().
Chromobox (Cbx) proteins contain a chromatin organizer modifier domain (chromodomain). The Prc1 member Cbx proteins, namely Cbx2, Cbx4, and Cbx8Citation39 were consistently expressed at low levels throughout liver development, whereas Cbx6 mRNA decreased but Cbx7 mRNA increased during postnatal development (). There are 3 mammalian homologs of Drosophila heterochromatin protein 1 (HP1) genes, namely Cbx5 (encoding Hp1α), Cbx1 (encoding Hp1β), and Cbx3 (encoding Hp1γ).Citation40 Hepatic mRNA expression of Cbx1/Hp1β, Cbx3/Hp1γ, and Cbx5/Hp1α decreased sharply from 2 d before birth (Day-2) to birth (Day0), and tended to further decrease after birth, resulting in low expression levels in adulthood (). In adult tissues, Cbx1, Cbx3, and Cbx5 mRNAs were 1–3 fold higher in kidney and small intestine than liver, Cbx2, Cbx4, and Cbx8 mRNA levels were comparable among the 3 tissues, whereas Cbx6 mRNAs were much (7–15 fold) higher in small intestine and kidney than liver ().
Ontogeny and tissue distribution of mRNAs of genes involved in histone demethylation
The lysine/histone demethylase (Kdm) family member Kdm1a and Kdm1b are H3K4 demethylases. Kdm1a, also known as lysine-specific demethylase 1 (Lsd1), demethylates mono- and dimethylated histone H3K4 and is essential for embryonic development.Citation41 Kdm1b, also known as Lsd2, is a histone H3K4 demethylase required to establish maternal genomic imprints.Citation42 Interestingly, hepatic Lsd1 mRNA decreased, whereas Lsd2 mRNA increased during postnatal development (). Kdm2a, also known as JmjC domain-containing histone demethylase 1a (Jhdm1a), recognizes CpG island and catalyzes the removal of H3K36 methylation, resulting in CpG island chromatin uniquely depleted of H3K36 methylation.Citation43 Kdm2b (also known as Jhdm1b) is also a H3K36 demethylase.Citation44 Hepatic Kdm2a mRNA fluctuated, whereas Kdm2b mRNA markedly decreased during postnatal development (). Kdm3a (also known as Jhdm1a/Jmjd1a) is a H3K9-specific demethylase important in regulating metabolic gene expression and obesity resistance.Citation45 Kdm3b (also known as JHhdm2b/Jmjd1b) specifically demethylates mono- and dimethyl-H3K9.Citation46 Kdm4a (also known as Jhdm3a/Jmjd2a) catalyzes the demethylation of di- and trimethylated K9 and K36 in histone H3.Citation47 Kdm4b (also known as Jmjd2b) antagonizes H3K9me3 at pericentric heterochromatin in mammalian cells via mediating the removal of H3K9me3.Citation48 Hepatic Kdm3a, Kdm3b, Kdm4a, and Kdm4b mRNAs decreased moderately (~50%) during postnatal development (). Kdm4c (also known as Jmjd2c/Jhdm3c) is preferentially expressed in undifferentiated ES cells.Citation49 Kdm4c mRNA was low, whereas the testis-enriched Kdm4d was undetectable throughout liver development (data not shown). Kdm5a (also known as Jarid1a/Rbp2), Kdm5b (also known as Jarid1b/Plu-1) and Kdm5c (also known as Jarid1c/Smcx) are specific for H3K4me3/me2 demethylation.Citation50 Hepatic Kdm5a mRNA decreased during postnatal development, but increased again in adulthood, whereas hepatic Kdm5b mRNA fluctuated during liver development (). Kdm5c mRNA expression was much higher than Kdm5a and Kdm5b during perinatal period, decreased 66% 10 d after birth, and remained stable thereafter (). In contrast, Kdm5d (Jarid1d), the last family member of Jarid1, was expressed lowly throughout liver development. Kdm6a/Utx and Kdm6b/Jmjd3 catalyze specifically the demethylation of H3K27me2/3.Citation16 Kdm7a/Jhdm1d is a dual demethylase for H3K9 and H3K27 and functions in brain development.Citation51 Hepatic Kdm6a/Utx mRNA was the highest before birth and decreased to 36% of pre-natal level in adulthood, whereas Kdm6b/Jmjd3 mRNA remained little changed during liver development (). Hepatic Kdm7a mRNA sharply decreased 70% from 2 d before birth to birth, and remained low thereafter ().
Figure 6. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in histone demethylation in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
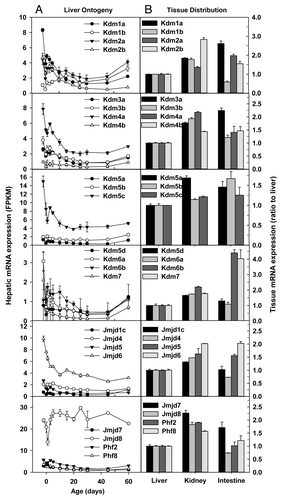
JMJD1C specifically demethylates histone H3K9 mono- and di-methylation,Citation52 whereas the function of Jmjd4 remains to be characterized. Hepatic Jmjd1c and Jmjd4 mRNAs were consistently low throughout liver development (). In contrast, Jmjd5/Kdm8, a H3K36me2 demethylase,Citation53 and Jmjd6, a histone arginine demethylaseCitation54 were expressed at 2–3 fold higher levels on Day-2 than adulthood (). The functions of Jmjd7 and Jmjd8 remain unknown. Hepatic Jmjd7 mRNA was 1.5 fold higher on Day-2 than adulthood, whereas Jmjd8 mRNA remained little changed during liver development (). PHD finger protein 2 (Phf2) and Pnf8 are mono-methyl histone H4 lysine 20 (H4K20me1) demethylases.Citation55 Hepatic Phf2 and Phf8 mRNAs were ~50% lower in adulthood than prenatally ().
In adulthood, the mRNA expression of the Kdm family members was generally higher in kidney and small intestine than liver, with the exception of Kdm1b, whose expression in small intestine was only 60% of that in liver ().
Ontogeny and tissue distribution of mRNAs of genes involved in histone arginine methylation and histone phosphorylation
In addition to lysines, the arginine residues within histones and other proteins can be methylated by a group of proteins known as protein arginine methyltransferases (Prmt).Citation56 Prmt1 methylates histone H4 at arginine 3, generating the activation mark of H4R3me2a. Hepatic Prmt1 mRNA on Day-2 was the highest among the Prmts, and its expression gradually decreased during postnatal development (). Prmt2 can methylate histone H4 and function as a transactivator. Prmt3 appears to be located exclusively in the cytosol and thus may not directly impact histones. Prmt2 and Prmt3 mRNAs were consistently low throughout liver development (). Prmt4, also known as Carm1, methylates histone H3 and does not share substrate with Prmt1. Prmt4/Carm1 functions as a transactivator and is essential for postnatal survival in mice. Prmt5 is a strong repressor of gene expression and it methylates histone H2A, H3, and H4. Prmt6 is the primary enzyme responsible for H3R2 methylation in mammalian cells, which counteracts the H3K4me3 activation mark, resulting in transcriptional repression. Prmt6 mRNAs was consistently low throughout liver development. Prmt7 appears to have different methylation activities toward different substrates. Hepatic mRNA expression of Prmt4/Carm1, Prmt5, and Prmt7 gradually decreased during postnatal development (). Prmt8 has a high degree of sequence identity with Prmt1, and its expression is restricted to the brain; Prmt8 mRNA was undetectable throughout liver development (data not shown). In adulthood, Prmt mRNAs were generally expressed at higher levels in kidney and small intestine than liver. Particularly, Prmt2 mRNAs were 11–16 fold higher in adult kidney and small intestine than adult liver ().
Figure 7. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in methylation of histone arginine residues and phosphorylation of histones in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
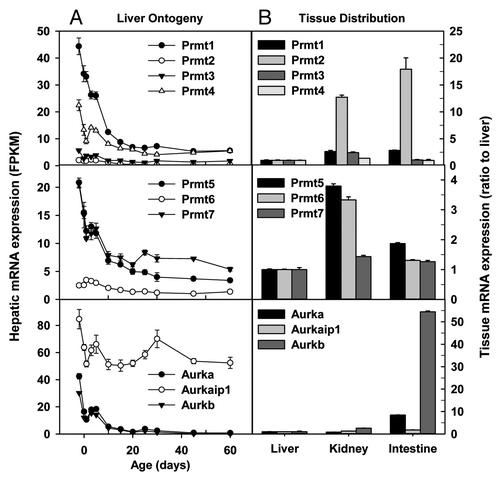
In addition to acetylation and methylation, phosphorylation is another important post-translational modification of histones.Citation57 Aurora kinases (Aurk) are essential for the phosphorylation of histone H3 at Ser10 and Ser28 during mitosis.Citation58 Aurka and Aurkb mRNAs were expressed at high levels in prenatal liver and markedly decreased during postnatal development (). In contrast, Aurka interacting protein 1 (Aurkaip1), which degrades Aurka through a proteasome-dependent pathway, was consistently expressed at high levels during liver development (). The mRNA expression of Aurkaip1 was similar in adult liver, kidney, and small intestine, whereas adult small intestine had 7- and 53-fold higher Aurka and Aurkb mRNAs, respectively, than adult liver ().
Ontogeny and tissue distribution of mRNAs of genes involved in chromosome remodeling
Chromosome remodeling refers to dynamic structural changes to the chromatin occurring throughout the cell division cycle. These changes range from the local changes necessary for transcriptional regulation to global changes necessary for chromosome segregation. Various chromosome remodeling factors have been identified. Smarca2/Brm, Smarca4/Brg1, and Smarcb1/Snf5 are key components within the SWI/SNF chromatin-remodeling complex, which plays essential roles in a variety of cellular processes including differentiation and proliferation.Citation59 Hepatic Brm mRNA gradually increased, whereas Brg1 and Snf5 mRNAs gradually decreased with postnatal development (). Interestingly, adult kidney and small intestine had ~1.7 fold higher and ~60% lower Brm mRNAs than adult liver, respectively (). In contrast, Brg1 and Snf5 mRNAs were expressed at 1–2 fold higher levels in adult kidney and small intestine than adult liver ().
Figure 8. Hepatic ontogeny (A) and tissue distribution (B) of mRNAs of genes involved in chromosome remodeling in male C57BL/6 mice. (A) Liver from C57BL/6 mice of ages from Day 2 to Day 60. n = 3, mean ± SE. Y-axis represents mRNAs expressed as fragments per kilobase of exon per million reads mapped (FPKM). (B) Liver, kidney, and small intestine from C57BL/6 mice of 60 d old. n = 2, mean ± SE. Y-axis represents relative mRNA expression with values of liver set as 1.0.
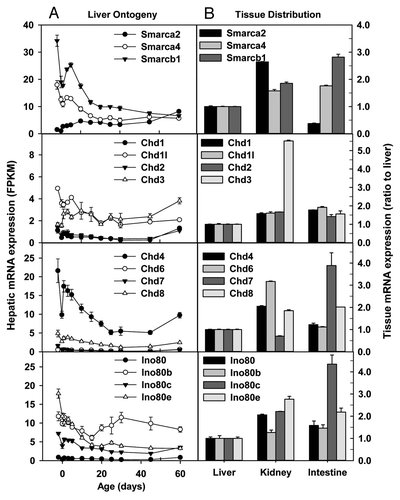
The Chromodomain Helicase DNA-binding (CHD) family of chromosome remodelers includes a number of proteins highly conserved from yeast to humans, though the functions of many of these proteins remain unknown or poorly characterized.Citation60 Hepatic mRNA expression of Chd1, Chd2, and Chd6 fluctuated at low levels throughout liver development (). Hepatic Chd1l, Chd7, and Chd8 mRNAs were ~1 fold higher on Day-2 than adulthood. Chd4/Mi-2b is required for the self-renewal of hematopoietic stem cells.Citation61 Hepatic Chd4 mRNA was much higher than other Chd family members on Day-2; Chd4 expression decreased gradually during postnatal development, and still remained higher than other Chd family members in adulthood (). Interestingly, hepatic Chd3 mRNA in adulthood was twice that on Day-2 (). In adulthood, Chd1, Chd1l1, Chd2, and Chd3 mRNAs were slightly higher in kidney and small intestine than liver, except that Chd3 mRNA in kidney was 4.5-fold higher than that in liver (). Additionally, Chd4, Chd6, and Chd8 mRNAs in adult kidney and small intestine tended to be moderately higher than liver. In contrast, Chd7 mRNA in kidney was slightly lower, whereas that in small intestine was ~3 fold higher than liver ().
The evolutionarily conserved INO80 family of ATP-dependent chromatin-remodeling enzymes has roles in many nuclear processes, particularly in the deposition and removal of the unacetylated histone variant H2A.Z.Citation62 Hepatic Ino80, Ino80b, and Ino80d mRNAs fluctuated, whereas Ino80c and Ino80e mRNAs decreased during postnatal liver development (). In adulthood, Ino80 family members tended to be expressed at higher levels in kidney and small intestine than liver ().
Real-time PCR validation of RNA-seq data
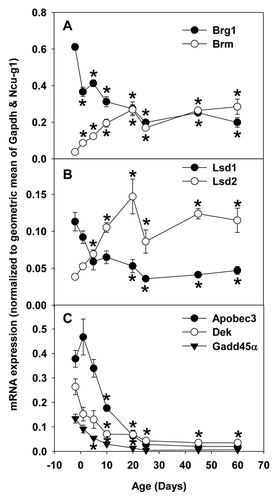
Consistent with RNA-seq data ( and ), real-time PCR data showed that hepatic mRNA expression of Brg1/Smarca4 () and Lsd1/Kdm1a () decreased moderately, whereas those of Brm/Smarca2 () and Lsd2/Kdm1b () increased markedly during postnatal development. In contrast, hepatic mRNA expression of Apobec3, Dek, and Gadd45α decreased sharply during postnatal development (), similar to the RNA-seq data ( and ).
Figure 9. Real-Time PCR validation of RNA-seq data of ontogenic mRNA expression of epigenetic factors in livers of male C57BL/6 mice. Total RNAs from livers of male C57BL/6 mice of ages of Day 2, 1, 5, 10, 20, 25, 45, and 60 were used for Real-time PCR validation. n = 3, mean ± SE. Y-axis represents mRNAs calculated by the comparative CT method, which determines the amount of target normalized to the geometric mean of Gapdh and Ncu-g1. * p < 0.05 vs. Day 2.
Discussion
The present study, for the first time, provide comprehensive analysis of developmental regulation of mRNA expression of various epigenetic factors, including enzymes involved in DNA methylation/demethylation, histone acetylation/deacetylation, histone methylation/demethylation, histone phosphorylation, and chromosome remodelers in mouse liver. Many of these epigenetic modifiers display ontogenic changes in mRNA expression in liver, with a vast majority of them downregulated during postnatal liver development. These epigenetic modifiers are generally expressed at higher levels in adult kidney and small intestine than adult liver, without marked tissue-specific expression patterns.
The present data indicate that the expression of many epigenetic modifiers is developmental-stage dependent rather than tissue-specific. Epigenetic modifications play a dominant role in development. Consistently, many epigenetic enzymes and chromosome remodelers are expressed at much higher levels in perinatal liver than adult liver. Different from transcription factors, which often demonstrate tissue-restricted expression patterns, the mRNA expression of the vast majority of epigenetic enzymes and chromosome remodelers do not have tissue-restricted expression patterns in the 3 adult tissues of liver, kidney, and small intestine. This is consistent with the general belief that tissue-specific/enriched transcription factors recruit general epigenetic enzymes to add/remove specific activating or silencing epigenetic modifications to the chromatin, resulting in tissue-specific patterns of gene expression.Citation1,Citation63
It is important to identify factors that determine the susceptibility of various organism/tissues to epigenetic modifiers. In adult liver, there is little cell proliferation, and the hepatocytes stay at G0. In contrast, cells in adult kidney and small intestine are more heterogeneous; a significant portion of stem cells/progenitor cells exist in adult kidney and small intestine.Citation64 Therefore, the mechanism of the observed moderately (~1–3 fold) higher mRNA expression of many epigenetic modifiers in adult kidney and/or small intestine vs. adult liver is, albeit unknown, likely due to the presence of higher amounts of stem cells/progenitor cells in these tissues. The present data suggest that in adulthood, kidney and small intestine are likely more susceptible to disturbance of gene expression by epigenetic modifiers. In contrast, the perinatal liver has high expression of epigenetic enzymes and undergo dramatic chromosome remodeling, making the developing liver highly susceptible to disturbance of epigenome by certain chemicals.
Physiological functions change dramatically from fetal liver to adult liver. Adult liver is the major organ to metabolize amino acids and lipids, to maintain gluconeogenesis, to synthesize serum proteins, and to detoxify xenobiotics. In contrast, fetal liver has fewer metabolic functions and is a major organ for hematopoiesis. Accordingly, many epigenetic enzymes important in hematopoiesis, such as Dnmt1,Citation65 Gadd45α,Citation66 Dot1l,Citation32 Ezh2,Citation67 Suv39h1,Citation68 and DekCitation69 are expressed highly in perinatal livers but minimally in adult livers. Interestingly, oncogenes DNMT1, EZH2, and DEK are overexpressed in human liver cancer.Citation70,Citation71 Apobec3, whose role in early liver development and hematopoisis remains unclear, is highly expressed in fetal liver, substantially downregulated in adult liver (Fig. One and 9), and widely overexpressed in human liver cancer.Citation72 In contrast, Gadd45α, a tumor-suppressorCitation73 expressed highly in fetal liver (Fig. One and 9), is downregulated in human liver cancer.Citation74 Understanding the regulatory mechanisms of ontogenic changes in mRNA expression of these important epigenetic modifiers will help understand the mechanisms of their overexpression/downregulation during liver carcinogenesis.
Interestingly, the mRNA expression of certain epigenetic modifiers increases with postnatal liver development, namely Brm, Lsd2, and Cbx7. Expression of the chromosome remodeler Brm is associated with cell differentiation, and Brm is downregulated in cancer.Citation75 Cbx7, a PcG protein and a tumor-suppressor, extends the lifespan of a wide range of normal human cells,Citation76 and its expression is downregulated in various types of cancer in humans.Citation77 LSD2/KDM1b/AOF1 is an H3K4me1/2 demethylase that specifically regulates histone H3K4 methylation within intragenic regions of its target genes; LSD2 associates predominantly with the gene bodies of actively transcribed genes, but is markedly absent from promoters.Citation78 The roles of Brm, Lsd2, and Cbx7 in regulating the epigenome, transcriptome, liver function, and hepatocarcinogenesis warrant further investigation.
It is intriguing to correlate the expression of epigenetic enzymes with the corresponding epigenetic signatures during liver development. The marked postnatal downregulation of Ezh2, a key enzyme for H3K27me3, correlates well with decreased H3K27me3 during tissue maturation.Citation79 Similarly, hepatic downregulation of Dnmts in mice is consistent with postnatal decreases in global DNA methylation in adult livers in humans.Citation79
It is noteworthy that only the mRNA levels of these epigenetic modifiers were investigated in this study. The gradual decrease in Brg1 mRNA but increase in Brm mRNA during postnatal liver development in mice () is consistent with the gradual decrease in Brg1 protein but increase in Brm protein during postnatal liver development in rats.Citation80 Additionally, the high and very low mRNA expression of Ncoa4 in fetal and adult mouse livers, respectively (), is consistent with intense and weak staining of Ncoa4 proteins in fetal and adult mouse livers, respectively.Citation81 More studies are needed to determine whether the marked changes in mRNA expression of many epigenetic modifiers during liver development translate into ontogenic alterations in their protein expression.
In summary, the present study provide the first knowledge about the ontogenic mRNA expression patterns of all major known epigenetic modifiers during mouse liver development and the distribution of these mRNAs among the 3 major tissues of drug processing, namely liver, kidney, and small intestine in adulthood. Such knowledge will serve as the foundation for further study on the epigenetic regulation of gene expression during liver development, as well as the epigenetic mechanism of tissue-specific gene expression in these tissues. Moreover, understanding the regulatory mechanisms of ontogenic changes of these epigenetic modifiers during liver development will also greatly aid the elucidation of the mechanisms of aberrant up- or downregulation of these epigenetic modifiers in liver cancer.
Materials and Methods
Animals
Eight-week old C57BL/6 breeding pairs of mice were purchased from The Jackson Laboratory. They were housed according to the American Animal Association Laboratory Animal Care guidelines, and were bred under standard conditions at the University of Kansas Medical Center. Livers from offspring were collected at the following 12 ages: day -2, 0 (right after birth and before the start of suckling), 1, 3, 5, 10, 15, 20, 25, 30, 45, and 60. Due to potential variations caused by the estrous cycle in maturing adult female mice, only male livers were used for this study (n = 3 per age, randomly selected from multiple litters). For the study of tissue distribution, tissues of liver, kidney, and small intestine were collected from adult male mice at 60 d of age (n = 2 per tissue). Tissues were frozen immediately in liquid nitrogen, and stored at -80°C before use.
RNA isolation
Total RNA from liver, kidney, and small intestine was isolated using RNAzol Bee reagent (Tel-Test Inc., Friendswood, TX) per the manufacturer’s protocol. RNA concentrations were quantified using a NanoDrop Spectrophotometer (NanoDrop Technologies, Wilmington, DE) at a wavelength of 260 nm. Integrity of the total RNA samples was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA) and the samples with RNA integrity values above 7.0 were used for the following experiments.
cDNA library preparation and RNA-Sequencing of liver ontogeny samples
The cDNA libraries from all total RNA samples of livers of 12 ages of mice were prepared using an Illumina TruSeq RNA sample prep kit (Illumina). Three micrograms of total RNA were used per the RNA input recommendations of the manufacturer’s protocol. The mRNAs were selected from the total RNAs by purifying the poly-A containing molecules using poly-T primers. The RNA fragmentation, first and second strand cDNA syntheses, end repair, adaptor ligation, and PCR amplification were performed per the manufacturer’s protocol. Fragments of the cDNA library ranged from 220 to 500 bps with an average size at 280 bp (including 100 bp adaptor sequences). The quality of cDNA libraries was validated using an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA) before sequencing by Genome Sequencing Facility at the University of Kansas Medical Center. Briefly, the cDNA libraries clustered onto a TruSeq paired-end flow cell and sequenced (2 × 100) using a TruSeq 200 cycle SBS kit (Illumina, San Diego, CA). For the initial run, a PhiX control was loaded on each flowcell as well as universal human reference RNA on 1 of the 16 lanes of the Illumina HiSeq2000 sequencer, and sequenced in parallel with other samples to ensure the data generated for each run are accurately calibrated during the image analysis and data analysis. In addition, the PhiX was spiked into each cDNA sample at approximately 1% as an internal quality control.
RNA-seq data analysis
After the sequencing platform generated the sequencing images, the pixel-level raw data collection, image analysis, and base calling were performed by the RTA (Real Time Analysis) software on a Dell PC attached to the HiSeq2000 sequencer. The data were streamed off to the analysis server as the run was progressing. The BCL Converter converted the BCL (base calling) files to qseq files, and the qseq files were subsequently converted to FASTQ files for downstream analysis. The RNA-seq reads from the FASTQ files were mapped to the mouse reference genome (NCBI37/mm9) and the splice junctions were identified by TopHat. The output files in BAM (binary sequence alignment) format were analyzed by Cufflinks to estimate the transcript abundance. The transcript structure predictions of Cufflinks are compared with Ensembl GTF version 65 by Cuffcompare. RNA-seq generated an average of 175 million reads per sample, and more than 80% of the reads were mapped to the mouse reference genome (NCBI37/mm9) by TopHat (data not shown). The mRNA abundance of various epigenetic modifiers was calculated by Cufflinks. For the study of liver ontogeny, the mRNA abundance was expressed in FPKM (fragments per kilobase of exon per million reads mapped), which normalizes sequencing depths between different samples and sizes between different genes, allowing direct comparison of expression levels among different transcripts in a genome-wide scale.
cDNA library preparation and RNA-Sequencing of tissue distribution samples
The preparation of cDNA libraries of adult male liver, kidney, and small intestine and sequencing of these cDNA libraries were conducted by Beijing Genomic Institute (BGI). Beads with oligo(dT) were used to isolate poly(A) mRNA from total RNA. Fragmentation buffer was added for interrupting mRNA to short fragments. Taking these short fragments (200–700 nucleotides) as templates, random hexamer primers were used to synthesize the first-strand cDNA. The second-strand cDNA was synthesized using buffer, dNTPs, RNase H and DNA polymerase I, respectively. Short fragments were purified with QiaQuick PCR extraction kit and resolved with EB buffer for end reparation and adding poly(A). After that, the short fragments were connected with sequencing adaptors. For amplification with PCR, suitable fragments were selected as templates with respect to the result of agarose gel electrophoresis. The resultant cDNA libraries were sequenced using Illumina HiSeq™ 2000. Images generated by sequencers were converted by base calling into nucleotide sequences, which were called raw reads and were stored in FASTQ format. After removal of dirty reads which contain adapters, unknown, or low quality bases from the raw reads, the resultant clean reads were mapped to reference mouse genome (NCBI37/mm9) and gene sequences respectively using SOAP2.Citation82 FPKM method was used to calculate mRNA abundance, which normalizes sequencing depths between different samples and sizes between different genes. For the study of tissue distribution, the mRNA abundance was expressed as ratio to value of liver, with that of liver set as 1.0.
Validation of RNA-seq data with real-time PCR
The total RNAs of male mouse livers at 8 ages of day -2, 1, 5, 10, 20, 25, 45, and 60 (n = 3 per age) were reverse-transcribed into cDNAs using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNAs were used for real-time PCR quantification of mRNA expression of Brg1, Brm, Lsd1, Lsd2, Apobec3, Dek, and Gadd45α using the iTaq™ Universal SYBR® Green Supermix and a MyiQ2 Real-Time PCR Detection System (Bio-Rad Laboratories). No single commonly-used housekeeping gene had consistent mRNA expression levels throughout liver development (RNA-seq data, not shown). In contrast, the average expression levels of two housekeeping genes, namely Gapdh and Ncu-g1 (an integral membrane protein of the lysosome), were found to be consistent throughout liver development (Fig. S3). Therefore, amounts of mRNAs of epigenetic factors were calculated using the comparative CT method, which determines the amount of target normalized to the geometric mean of Gapdh and Ncu-g1. Real-Time PCR primers are listed in Table S2.
Statistics
Data are presented as mean ± standard error (SE). Differences between various groups were determined using ANOVA followed by post-hoc test, with significance set at p < 0.05. In the study of liver ontogeny, the FPKM values were log2 transformed to achieve normal distribution prior to ANOVA. The statistics (p values) of mRNA expression of epigenetic modifiers during liver development is shown in Table S1.
| Abbreviations: | ||
| Apobec | = | activation-induced cytidine deaminase/(AID)/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like |
| Aurk | = | aurora kinases |
| Chd | = | chromodomain helicase DNA-binding |
| Dnmt | = | DNA methyltransferase |
| Ehmt | = | euchromatin histone methyltransferase |
| Ezh | = | enhancer of zeste homolog |
| FPKM | = | fragments per kilobase of exon per million reads mapped |
| Gadd45 | = | growth arrest and DNA-damage-inducible protein 45 |
| Hdac | = | histone deacetylase |
| Hp1 | = | heterochromatin protein 1 |
| Kat | = | lysine acetyltransferase |
| Kdm | = | lysine/histone demethylase |
| Lsd | = | lysine-specific demethylase |
| Mbd | = | methyl-CpG binding domain protein |
| PcG | = | Polycomb group |
| Prc | = | polycomb repressive complex |
| Prmt | = | protein arginine methyltransferases |
| SetD | = | SET domain containing |
| Smyd | = | SET and MYND domain containing |
| Tet | = | ten-eleven translocation |
Additional material
Download Zip (122.4 KB)Acknowledgments
We thank Helen Renaud and Lai Peng for technical assistance in animal experiments as well as Clark Bloomer from the KUMC Sequencing Core Facilities for his technical assistance in mRNA-seq. We thank NIH grants ES019487 and RR021940 for financial support.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Snykers S, Henkens T, De Rop E, Vinken M, Fraczek J, De Kock J, et al. Role of epigenetics in liver-specific gene transcription, hepatocyte differentiation and stem cell reprogrammation. J Hepatol 2009; 51:187 - 211; http://dx.doi.org/10.1016/j.jhep.2009.03.009; PMID: 19457566
- Klaassen CD, Lu H, Cui JY. Epigenetic regulation of drug processing genes. Toxicol Mech Methods 2011; 21:312 - 24; http://dx.doi.org/10.3109/15376516.2011.562758; PMID: 21495869
- Imai S, Kikuchi R, Kusuhara H, Yagi S, Shiota K, Sugiyama Y. Analysis of DNA methylation and histone modification profiles of liver-specific transporters. Mol Pharmacol 2009; 75:568 - 76; http://dx.doi.org/10.1124/mol.108.052589; PMID: 19047482
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21; http://dx.doi.org/10.1101/gad.947102; PMID: 11782440
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 2005; 74:481 - 514; http://dx.doi.org/10.1146/annurev.biochem.74.010904.153721; PMID: 15952895
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693 - 705; http://dx.doi.org/10.1016/j.cell.2007.02.005; PMID: 17320507
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007; 130:77 - 88; http://dx.doi.org/10.1016/j.cell.2007.05.042; PMID: 17632057
- Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I, et al. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res 2009; 19:221 - 33; http://dx.doi.org/10.1101/gr.080861.108; PMID: 19047520
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005; 120:169 - 81; http://dx.doi.org/10.1016/j.cell.2005.01.001; PMID: 15680324
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, et al. A high-resolution map of active promoters in the human genome. Nature 2005; 436:876 - 80; http://dx.doi.org/10.1038/nature03877; PMID: 15988478
- Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A 2006; 103:15782 - 7; http://dx.doi.org/10.1073/pnas.0607617103; PMID: 17043231
- Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev 2005; 19:542 - 52; http://dx.doi.org/10.1101/gad.1272505; PMID: 15706033
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006; 441:349 - 53; http://dx.doi.org/10.1038/nature04733; PMID: 16625203
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006; 125:301 - 13; http://dx.doi.org/10.1016/j.cell.2006.02.043; PMID: 16630818
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Swigut T, Wysocka J. H3K27 demethylases, at long last. Cell 2007; 131:29 - 32; http://dx.doi.org/10.1016/j.cell.2007.09.026; PMID: 17923085
- Sims RJ 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet 2003; 19:629 - 39; http://dx.doi.org/10.1016/j.tig.2003.09.007; PMID: 14585615
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell 2007; 128:707 - 19; http://dx.doi.org/10.1016/j.cell.2007.01.015; PMID: 17320508
- Krauss V. Glimpses of evolution: heterochromatic histone H3K9 methyltransferases left its marks behind. Genetica 2008; 133:93 - 106; http://dx.doi.org/10.1007/s10709-007-9184-z; PMID: 17710556
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 2009; 41:246 - 50; http://dx.doi.org/10.1038/ng.297; PMID: 19151716
- Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001; 410:116 - 20; http://dx.doi.org/10.1038/35065132; PMID: 11242053
- Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol 2011; 9:34; http://dx.doi.org/10.1186/1741-7007-9-34; PMID: 21627854
- Cui JY, Gunewardena SS, Yoo B, Liu J, Renaud HJ, Lu H, et al. RNA-Seq Reveals Different mRNA Abundance of Transporters and Their Alternative Transcript Isoforms During Liver Development. Toxicol Sci 2012; 127:592 - 608; http://dx.doi.org/10.1093/toxsci/kfs107; PMID: 22454430
- Peng L, Yoo B, Gunewardena SS, Lu H, Klaassen CD, Zhong XB. RNA Sequencing Reveals Dynamic Changes of mRNA Abundance of Cytochromes P450 and Their Alternative Transcripts during Mouse Liver Development. Drug Metab Dispos 2012; 40:1198 - 209; http://dx.doi.org/10.1124/dmd.112.045088; PMID: 22434873
- Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 2009; 8:1526 - 31; http://dx.doi.org/10.4161/cc.8.10.8500; PMID: 19377292
- Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell 2011; 146:866 - 72; http://dx.doi.org/10.1016/j.cell.2011.08.042; PMID: 21925312
- Voss AK, Thomas T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays 2009; 31:1050 - 61; http://dx.doi.org/10.1002/bies.200900051; PMID: 19722182
- Xu J, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer 2009; 9:615 - 30; http://dx.doi.org/10.1038/nrc2695; PMID: 19701241
- Shao W, Halachmi S, Brown M. ERAP140, a conserved tissue-specific nuclear receptor coactivator. Mol Cell Biol 2002; 22:3358 - 72; http://dx.doi.org/10.1128/MCB.22.10.3358-3372.2002; PMID: 11971969
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370:737 - 49; http://dx.doi.org/10.1042/BJ20021321; PMID: 12429021
- Steward MM, Lee JS, O’Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol 2006; 13:852 - 4; http://dx.doi.org/10.1038/nsmb1131; PMID: 16892064
- Feng Y, Yang Y, Ortega MM, Copeland JN, Zhang M, Jacob JB, et al. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 2010; 116:4483 - 91; http://dx.doi.org/10.1182/blood-2010-03-276501; PMID: 20798234
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 2005; 19:815 - 26; http://dx.doi.org/10.1101/gad.1284005; PMID: 15774718
- Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res 2011; 17:2613 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-10-2156; PMID: 21367748
- Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schöfer C, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001; 107:323 - 37; http://dx.doi.org/10.1016/S0092-8674(01)00542-6; PMID: 11701123
- O’Carroll D, Scherthan H, Peters AH, Opravil S, Haynes AR, Laible G, et al. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol 2000; 20:9423 - 33; http://dx.doi.org/10.1128/MCB.20.24.9423-9433.2000; PMID: 11094092
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 2004; 18:1251 - 62; http://dx.doi.org/10.1101/gad.300704; PMID: 15145825
- Kappes F, Waldmann T, Mathew V, Yu J, Zhang L, Khodadoust MS, et al. The DEK oncoprotein is a Su(var) that is essential to heterochromatin integrity. Genes Dev 2011; 25:673 - 8; http://dx.doi.org/10.1101/gad.2036411; PMID: 21460035
- Senthilkumar R, Mishra RK. Novel motifs distinguish multiple homologues of Polycomb in vertebrates: expansion and diversification of the epigenetic toolkit. BMC Genomics 2009; 10:549; http://dx.doi.org/10.1186/1471-2164-10-549; PMID: 19930571
- Zeng W, Ball AR Jr., Yokomori K. HP1: heterochromatin binding proteins working the genome. Epigenetics 2010; 5:287 - 92; http://dx.doi.org/10.4161/epi.5.4.11683; PMID: 20421743
- Foster CT, Dovey OM, Lezina L, Luo JL, Gant TW, Barlev N, et al. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol 2010; 30:4851 - 63; http://dx.doi.org/10.1128/MCB.00521-10; PMID: 20713442
- Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, et al. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009; 461:415 - 8; http://dx.doi.org/10.1038/nature08315; PMID: 19727073
- Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ. CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell 2010; 38:179 - 90; http://dx.doi.org/10.1016/j.molcel.2010.04.009; PMID: 20417597
- He J, Kallin EM, Tsukada Y, Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat Struct Mol Biol 2008; 15:1169 - 75; http://dx.doi.org/10.1038/nsmb.1499; PMID: 18836456
- Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009; 458:757 - 61; http://dx.doi.org/10.1038/nature07777; PMID: 19194461
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006; 125:483 - 95; http://dx.doi.org/10.1016/j.cell.2006.03.027; PMID: 16603237
- Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol 2007; 14:689 - 95; http://dx.doi.org/10.1038/nsmb1273; PMID: 17589523
- Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev 2006; 20:1557 - 62; http://dx.doi.org/10.1101/gad.388206; PMID: 16738407
- Katoh Y, Katoh M. Comparative integromics on JMJD2A, JMJD2B and JMJD2C: preferential expression of JMJD2C in undifferentiated ES cells. Int J Mol Med 2007; 20:269 - 73; PMID: 17611647
- Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, et al. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 2007; 128:1063 - 76; http://dx.doi.org/10.1016/j.cell.2007.02.003; PMID: 17320161
- Tsukada Y, Ishitani T, Nakayama KI. KDM7 is a dual demethylase for histone H3 Lys 9 and Lys 27 and functions in brain development. Genes Dev 2010; 24:432 - 7; http://dx.doi.org/10.1101/gad.1864410; PMID: 20194436
- Kim SM, Kim JY, Choe NW, Cho IH, Kim JR, Kim DW, et al. Regulation of mouse steroidogenesis by WHISTLE and JMJD1C through histone methylation balance. Nucleic Acids Res 2010; 38:6389 - 403; http://dx.doi.org/10.1093/nar/gkq491; PMID: 20530532
- Hsia DA, Tepper CG, Pochampalli MR, Hsia EY, Izumiya C, Huerta SB, et al. KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc Natl Acad Sci U S A 2010; 107:9671 - 6; http://dx.doi.org/10.1073/pnas.1000401107; PMID: 20457893
- Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science 2007; 318:444 - 7; http://dx.doi.org/10.1126/science.1145801; PMID: 17947579
- Qi HH, Sarkissian M, Hu GQ, Wang Z, Bhattacharjee A, Gordon DB, et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010; 466:503 - 7; http://dx.doi.org/10.1038/nature09261; PMID: 20622853
- Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS Lett 2011; 585:2024 - 31; http://dx.doi.org/10.1016/j.febslet.2010.11.010; PMID: 21074527
- Banerjee T, Chakravarti D. A peek into the complex realm of histone phosphorylation. Mol Cell Biol 2011; 31:4858 - 73; http://dx.doi.org/10.1128/MCB.05631-11; PMID: 22006017
- Pascreau G, Arlot-Bonnemains Y, Prigent C. Phosphorylation of histone and histone-like proteins by aurora kinases during mitosis. Prog Cell Cycle Res 2003; 5:369 - 74; PMID: 14593731
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011; 11:481 - 92; http://dx.doi.org/10.1038/nrc3068; PMID: 21654818
- Ho L, Crabtree GR. Chromatin remodelling during development. Nature 2010; 463:474 - 84; http://dx.doi.org/10.1038/nature08911; PMID: 20110991
- Yoshida T, Hazan I, Zhang J, Ng SY, Naito T, Snippert HJ, et al. The role of the chromatin remodeler Mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev 2008; 22:1174 - 89; http://dx.doi.org/10.1101/gad.1642808; PMID: 18451107
- Watanabe S, Peterson CL. The INO80 family of chromatin-remodeling enzymes: regulators of histone variant dynamics. Cold Spring Harb Symp Quant Biol 2010; 75:35 - 42; http://dx.doi.org/10.1101/sqb.2010.75.063; PMID: 21502417
- Stein GS, Zaidi SK, Stein JL, Lian JB, van Wijnen AJ, Montecino M, et al. Transcription-factor-mediated epigenetic control of cell fate and lineage commitment. Biochem Cell Biol 2009; 87:1 - 6; http://dx.doi.org/10.1139/O08-094; PMID: 19234518
- Orlando G, Baptista P, Birchall M, De Coppi P, Farney A, Guimaraes-Souza NK, et al. Regenerative medicine as applied to solid organ transplantation: current status and future challenges. Transpl Int 2011; 24:223 - 32; http://dx.doi.org/10.1111/j.1432-2277.2010.01182.x; PMID: 21062367
- Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009; 5:442 - 9; http://dx.doi.org/10.1016/j.stem.2009.08.016; PMID: 19796624
- Gupta SK, Gupta M, Hoffman B, Liebermann DA. Hematopoietic cells from gadd45a-deficient and gadd45b-deficient mice exhibit impaired stress responses to acute stimulation with cytokines, myeloablation and inflammation. Oncogene 2006; 25:5537 - 46; http://dx.doi.org/10.1038/sj.onc.1209555; PMID: 16732331
- Mochizuki-Kashio M, Mishima Y, Miyagi S, Negishi M, Saraya A, Konuma T, et al. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 2011; 118:6553 - 61; http://dx.doi.org/10.1182/blood-2011-03-340554; PMID: 22042701
- Czvitkovich S, Sauer S, Peters AH, Deiner E, Wolf A, Laible G, et al. Over-expression of the SUV39H1 histone methyltransferase induces altered proliferation and differentiation in transgenic mice. Mech Dev 2001; 107:141 - 53; http://dx.doi.org/10.1016/S0925-4773(01)00464-6; PMID: 11520670
- Broxmeyer HE, Kappes F, Mor-Vaknin N, Legendre M, Kinzfogl J, Cooper S, et al. DEK Regulates Hematopoietic Stem Engraftment and Progenitor Cell Proliferation. Stem Cells Dev 2011; PMID: 21943234
- Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol Res 2007; 37:974 - 83; http://dx.doi.org/10.1111/j.1872-034X.2007.00141.x; PMID: 17584191
- Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, et al. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res 1999; 59:4990 - 6; PMID: 10519413
- Xu R, Zhang X, Zhang W, Fang Y, Zheng S, Yu XF. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus B x antigen mutants and hepatocellular carcinoma. Hepatology 2007; 46:1810 - 20; http://dx.doi.org/10.1002/hep.21893; PMID: 17847074
- Rosemary Siafakas A, Richardson DR. Growth arrest and DNA damage-45 alpha (GADD45alpha). Int J Biochem Cell Biol 2009; 41:986 - 9; http://dx.doi.org/10.1016/j.biocel.2008.06.018; PMID: 18760377
- Gramantieri L, Chieco P, Giovannini C, Lacchini M, Treré D, Grazi GL, et al. GADD45-alpha expression in cirrhosis and hepatocellular carcinoma: relationship with DNA repair and proliferation. Hum Pathol 2005; 36:1154 - 62; http://dx.doi.org/10.1016/j.humpath.2005.07.017; PMID: 16260267
- Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene 2009; 28:1653 - 68; http://dx.doi.org/10.1038/onc.2009.4; PMID: 19234488
- Gil J, Bernard D, Martínez D, Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat Cell Biol 2004; 6:67 - 72; http://dx.doi.org/10.1038/ncb1077; PMID: 14647293
- Forzati F, Federico A, Pallante P, Abbate A, Esposito F, Malapelle U, et al. CBX7 is a tumor suppressor in mice and humans. J Clin Invest 2012; 122:612 - 23; http://dx.doi.org/10.1172/JCI58620; PMID: 22214847
- Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell 2010; 39:222 - 33; http://dx.doi.org/10.1016/j.molcel.2010.07.008; PMID: 20670891
- Brunner AL, Johnson DS, Kim SW, Valouev A, Reddy TE, Neff NF, et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res 2009; 19:1044 - 56; http://dx.doi.org/10.1101/gr.088773.108; PMID: 19273619
- Inayoshi Y, Miyake K, Machida Y, Kaneoka H, Terajima M, Dohda T, et al. Mammalian chromatin remodeling complex SWI/SNF is essential for enhanced expression of the albumin gene during liver development. J Biochem 2006; 139:177 - 88; http://dx.doi.org/10.1093/jb/mvj015; PMID: 16452305
- Kollara A, Brown TJ. Variable expression of nuclear receptor coactivator 4 (NcoA4) during mouse embryonic development. J Histochem Cytochem 2010; 58:595 - 609; http://dx.doi.org/10.1369/jhc.2010.955294; PMID: 20354146
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009; 25:1966 - 7; http://dx.doi.org/10.1093/bioinformatics/btp336; PMID: 19497933