Abstract
We have recently reported that in astrocytoma cells the expression of interleukin-8 (IL-8) is upregulated by prostaglandin E2 (PGE2). Unfortunately, the exact mechanism by which this happens has not been clarified yet. Here, we have investigated whether IL-8 activation by PGE2 involves changes in DNA methylation and/or histone modifications in 46 astrocytoma specimens, two astrocytoma cell lines and normal astrocytic cells. The DNA methylation status of the IL-8 promoter was analyzed by bisulphite sequencing and by methylation DNA immunoprecipitation analysis. The involvement of DNA methyltransferases (DNMTs) and histone deacetylases (HDACs), as well as histone acetylation levels, was assayed by chromatin immunoprecipitation. IL-8 expression at promoter, mRNA and protein level was explored by transfection, real-time PCR and enzyme immunoassay experiments in cells untreated or treated with PGE2, 5-aza-2'-deoxycytidine (5-aza-dC) and HDAC inhibitors, alone or in combination. EMSA was performed with crude cell extracts or recombinant protein. We observed that PGE2 induced IL-8 activation through: (1) demethylation of the single CpG site 5 located at position -83 within the binding region for CEBP-β in the IL-8 promoter; (2) C/EBP-β and p300 co-activator recruitment; (3) H3 acetylation enhancement and (4) reduction in DNMT1, DNMT3a, HDAC2 and HDAC3 association to CpG site 5 region. Treatment with 5-aza-dC or HDAC inhibitors of class I HDACs strengthened either basal or PGE2-mediated IL-8 expression. These findings have elucidated an orchestrated mechanism triggered by PGE2 whereby concurrent association of site-specific demethylation and histone H3 hyperacetylation resulted in derepression of IL-8 gene expression in human astrocytoma.
Introduction
Changes in DNA methylation status and alterations in chromatin structure by histone modification represent the major epigenetic mechanisms implicated in the regulation of gene transcription in mammals.Citation1-Citation3 Recent studies suggest that these alterations may be important in the process of neoplasia, depending on the target genes involved.Citation4-Citation6 Aberrant promoter methylation or inappropriate recruitment of histone deacetylases (HDACs) might repress the transcription of tumor suppressor genes, whereas DNA demethylation or histone hyperacetylation processes could activate proto-oncogenes. Emerging evidence suggests that these two epigenetic marks are strictly linked and can reciprocally associate or interfere.Citation7,Citation8 The comprehension of the role of these processes and of their reciprocal interaction in carcinogenesis is relevant because of their reversibility and the possibility of drug treatments in both processes. In several tumors, IL-8 expression, which is tightly regulated under physiological conditions, strongly increases after a wide range of stimuli. IL-8, which was initially known for its role in the recruitment and activation of immune and inflammatory cells, is now believed to favor the invasion and metastatic spread of cancer cells as well as to increase angiogenesis in growing tumors.Citation9,Citation10 Very recent research data support that overexpression of IL-8 promotes glial tumor neovascularity and invasiveness that it is associated with patients’ survival.Citation11-Citation13 As we have recently shown, the mRNA levels of PGES-1, which is highly expressed in a large number of astrocytoma samples, positively correlate with those of IL-8 and, in astrocytoma cells, exposure to PGE2 induces IL-8 gene transcription.Citation14 Here we aimed at evaluating whether epigenetic events play a critical role in the PGE2-induced transcriptional activation of IL-8 in astrocytoma. To this end, we investigated the functional relevance of DNA methylation status and histone modifications in the PGE2-dependent regulation of IL-8 gene expression. We found that: (1) PGE2 induces demethylation of a specific single CpG site located within the binding region for the transcription factor CEBP-β in the promoter-enhancer of the IL-8 gene by reducing DNMT1 and DNMT3a recruitment; (2) demethylation of this cytosine residue favors the binding of C/EBP-β to the promoter-enhancer region in association with the co-activator p300, leading to increased IL-8 expression; (3) PGE2 dramatically enhances H3 acetylation by dissociating HDAC2 and HDAC3 from the IL-8 promoter and (4) treatment with demethylating agent 5-aza-dC or class I-selective or non-class-selective HDAC inhibitors (HDACis) significantly raises either basal or PGE2-induced IL-8 transcription.
Results
IL-8 mRNA levels correlate with the methylation status of CpG site 5 in astrocytoma tumor samples
Forty-six astrocytic glioma specimens were examined in order to determine the methylation pattern of the six CpG sites in the IL-8 promoter and the relative IL-8 mRNA levels. CpG sites located at -1311 (site 1), -1241 (site 2), -168 (site 3), -158 (site 4), -83 (site 5) and -7 (site 6) from the transcription start site () were analyzed by using the sodium bisulfite sequencing technique. As shown in , evidence of methylation was observed in 11 (23.9%), 12 (26.1%), 20 (43.5%), 21 (45.6%), 23 (50%) and 21 (45.6%) tumoral samples for the sites 1, 2, 3, 4, 5 and 6, respectively.
Figure 1. CpG methylation patterns of IL-8 promoter and IL-8 mRNA and protein levels in astrocytoma specimens. (A) Sequence of the IL-8 upstream region. The IL-8 promoter contain 6 CpG sites (underlined) located at -1311 (site 1), -241 (site 2), -168 (site 3), -158 (site 4), -83 (site 5) and -7 (site 6) respect to IL-8 transcriptional starting site (arrow). (B) Detailed methylation analysis of the 6 CpG residues at the IL-8 promoter in 46 astrocytoma specimens was determined by PCR-based direct sequencing of bisulfite-treated DNA. White square, unmethylated CpG site; black and white square, heterozygous methylated CpG site; black square, homozygous methylated CpG site. (C) Real-Time PCR analysis of IL-8 gene expression levels. Total RNA was extracted, reverse-transcribed, and analyzed by quantitative Real Time-PCR. mRNA levels were normalized by using the housekeeping gene β-actin as the inner control. Data are depicted as the mean ± SD of three independent experiments. (D) The amount of IL-8 protein was measured by ELISA. Data are depicted as the mean ± SD of three independent experiments.
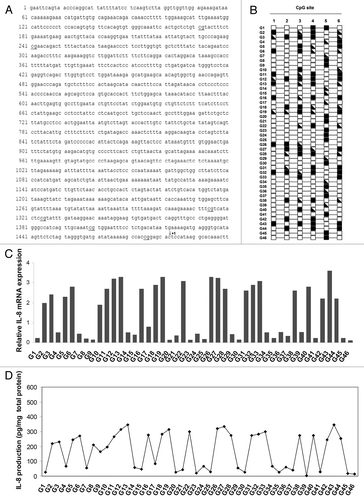
We analyzed astrocytoma samples for amount of IL-8 mRNA by real-time PCR to elucidate whether the methylation status affects IL-8 expression levels (). Twenty-three (50%) tumoral tissues showed a complete absence or very low levels of IL-8 expression. This reflected the response at the protein level, i.e., that there was a one-to-one correspondence of protein to mRNA (). A concordance between IL-8 mRNA expression and CpG site methylation was found in 12 (26.1%), 11 (23.9%), 3 (6.5%), 4 (8.7%), 46 (100%) and 4 (8.7%) tumoral samples for the sites 1, 2, 3, 4, 5 and 6, respectively. Pearson correlation analysis revealed that aberrant methylation of the individual CpG site 5 was significantly associated with IL-8 silencing (p < 0.0001). No significant association between the methylation pattern of the other CpG sites and the levels of IL-8 expression was found. From these observations we speculated that, in human astrocytoma, the methylation status of CpG site 5 in the IL-8 promoter is critical for IL-8 expression.
PGE2 induces demethylation at the CpG site 5 of IL-8 promoter in astrocytoma cells
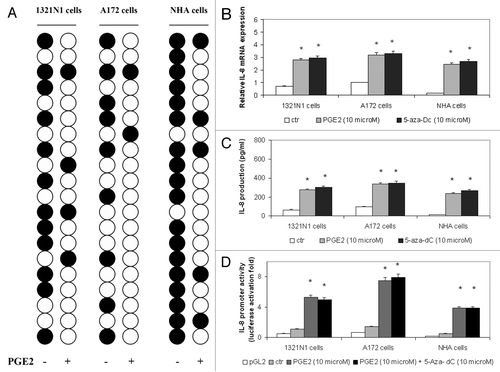
As we have previously shown that astrocytoma cells overexpressed IL-8 when stimulated with PGE2,Citation14 we tested the hypothesis that PGE2 may regulate IL-8 production through active DNA demethylation. To this aim, we determined the methylation status of IL-8 enhancer/promoter in 20 individual clones obtained from untreated or PGE2-treated 1321N1 and A172 astrocytoma cell lines and normal human astrocyte (NHA) cells. As shown in , CpG site 5 (nucleotide -83) was methylated in 60% of clones derived from 1321N1 cells, 45% of clones isolated from A172 cells and 90% of clones obtained from NHA cells, whereas it resulted to be largely unmethylated in PGE2-stimulated cells (80% in 1321N1 cells, 90% in A172 cells and 75% in NHA cells). In contrast, the methylation status of the other CpG sites was unmodified by PGE2 treatment (data not shown).
Figure 2. The PGE2-induced demethylation of CpG site 5 (nucleotide -83) in the IL-8 enhancer-promoter region increased IL-8 expression in astrocytoma cells. (A) 1321N1, A172 and NHA cells were stimulated with 10 µM PGE2 for 5 h. The methylation status of CpG site 5 was determined by bisulfite sequencing analysis in 20 individual clones from each cell line (white circle, unmethylated; black circle, methylated). (B) Cells were treated with PGE2 or 5-aza-dC. Total RNA was extracted, reverse-transcribed, and analyzed by quantitative real time-PCR. IL-8 mRNA levels were normalized by using the housekeeping gene β-actin as the inner control. Data are depicted as the mean ± SD of three independent experiments. (C) Cells were treated with PGE2 or 5-aza-dC. The amount of IL-8 protein was measured by ELISA. Data are depicted as the mean ± SD of three independent experiments. (D) Cells were transiently transfected with 1 µg of the IL-8 promoter construct and subsequently treated with PGE2 or 5-aza-dC. Data are expressed as mean ± SD of results in three independent experiments. Statistical analyses were performed compared with untreated control cells. *p < 0.001; absence of asterisks, not significant.
PGE2-induced demethylation at the CpG site 5 was associated with IL-8 reactivation
To investigate the role of the demethylation at CpG site 5 in the transcriptional regulation of the IL-8 gene, we examined the effects of either PGE2 or DNA methyltransferase inhibitor 5-aza-2'-deoxycytidine (5-aza-dC) on IL-8 at the mRNA, protein and promoter level in 1321N1, A172 and NHA cell lines. We found that 5-aza-dC increased IL-8 at mRNA (), protein () and promoter () level with an effect that can be compared with the activation induced by PGE2. These results suggest that demethylation at CpG site 5 reactivates IL-8 gene expression.
In vitro methylation of the IL-8 promoter at CpG site 5 abrogated the binding of C/EBP-β
Since the CpG site 5 lies within the cis-regulatory elements that bind the transcription factor C/EBP-β, we performed EMSA with an unmethylated (UnM) or methylated (M) probe spanning the C/EBP-β active site of the IL-8 enhancer. A strong complex was observed when nuclear extracts from 1321N1 astrocytoma cells stimulated with PGE2 were incubated with UnM-probe C/EBP-β (, lane 4). The band was supershifted by the antibody against C/EBP-β (, lane 5). In contrast, no nucleoprotein complex was observed when the M-probe C/EBP-β (probe B) was used (, lanes 6 and 7). The nuclear extracts from A172 and NHA cell lines showed a similar behavior (see Fig. S1). To provide evidence that the protein bound to the site was C/EBP-β, we performed EMSA analysis using a purified recombinant C/EBP-β protein. The interaction pattern was identical to the one obtained with crude cell extracts (, lanes 10–13). This suggests that C/EBP-β is indeed the protein that occupies the response element containing the CpG site 5 within the IL-8 promoter.
Figure 3. CpG site 5 methylation inhibits the binding of C/EBP-β to IL-8 promoter. Nuclear extracts prepared from untreated (lanes 3,6) or 10 µM PGE2-treated (lanes 4,7) 1321N1 cells, or recombinant C/EBP-β protein (lanes 10–13), were incubated with double-stranded oligonucleotide probe (-95/-52 bp) containing either unmethylated cytosine (UnM-probe C/EBP-β) or methylated cytosine (M-probe C/EBP-β) at CpG site 5 (nucleotide -83). In supershift analysis, 2 μg antibody against C/EBP-β (lanes 5,11,13) was incubated at room temperature (for 20 min) with the nuclear extracts prior to probe addition. Competition with a 200-fold excess of either unlabeled UnM-probe C/EBP-β (lane 8) or unlabeled M-probe C/EBP-β (lane 9) showed the specificity of protein binding.
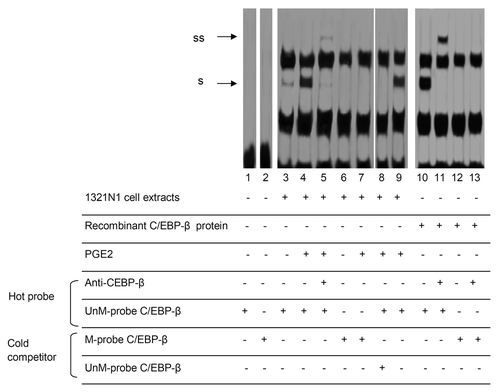
These results indicate that methylation of the CpG site 5 abrogates the binding of C/EBP-β to the IL-8 promoter-enhancer element.
Methylation of CpG site 5 of the IL-8 promoter downregulates IL-8 transcription
According to the EMSA results, it is likely that the methylation of CpG site 5, abrogating the binding of C/EBP-β transcription factor, also prohibits IL-8 gene transcription. To solve this issue, we methylated CpG site 5 using a promoter-directed siRNA method.Citation15 We constructed a short hairpin RNA (shRNA) targeted to the IL-8 promoter that included CpG site 5 in an expression vector and used it to transfect astrocytoma cells. Methylation of CpG site 5 was observed in 90% and 85% of sequences of 1321N1 and A172 cells, respectively (), paralleled by reduced IL-8 gene expression (). The other CpG sites were not affected (). We introduced IL-8 shRNA into astrocytoma cells in the presence of 5-aza-dC to verify whether the methylation at the specific cytosine residue 5 was necessary and sufficient to decrease IL-8 expression. Treatment with 5-aza-dC strongly reduced the DNA methylation at CpG site 5 (data not shown) and restored IL-8 expression (). In order to establish that CpG site 5 was involved in IL-8 transcription, we studied the effect of in vitro methylation of this site on IL-8 expression by using a reporter assay. These experiments showed that methylation of CpG site 5 completely disabled the ability of the promoter to direct the reporter gene expression, as shown in .
Figure 4. CpG site 5 methylation reduced IL-8 expression. (A) Induction of CpG site 5 methylation in genomic DNA using the RNAi method. 1321N1 and A172 cells were transfected with either control shRNA or target shRNA directed against CpG site 5 (IL-8 shRNA). The methylation status of the CpG site 5 was determined by bisulfite sequencing. (B) 1321N1 and A172 cells were transfected with either control shRNA or IL-8 shRNA in the presence or absence of 5-aza-dC. Expression levels of IL-8 mRNA were determined by Real-Time PCR. The results shown are the means ± SD of three independent experiments. (C) Demethylation of CpG site 5 is necessary for the enhancement of IL8-promoter-driven transcription. Luciferase activity produced by the indicated plasmids, either methylated (black circle) or unmethylated (white circle), was determined after their transfection into 1321N1 and A172 cells untreated (white boxes) or treated with 10 µM PGE2 (black boxes). Data are depicted as the mean ± SD of three independent experiments. Statistical analyses were performed compared with respective untreated control cells. *p < 0.01; absence of asterisks, not significant.
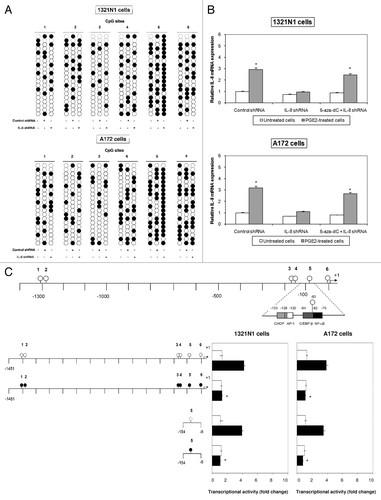
Combined, these findings indicate that the methylation status of the CpG site 5 is a crucial factor regulating IL-8 promoter transcription in astrocytoma cells.
PGE2 induces the binding of C/EBP-βand p300 to IL-8 promoter and histone H3 acetylation
Chromatin immunoprecipitation (ChIP) experiments were conducted to examine the in vivo accessibility of C/EBP-β to the IL-8 promoter region in the presence of PGE2. As it is shown in , in untreated cells, C/EBP-β binding to the IL-8 promoter was very weak, whereas, within 3 h from PGE2 treatment, the amount of endogenous C/EBP-β that occupied the IL-8 promoter in vivo was consistently higher. The interaction of C/EBP-β with IL-8 promoter peaked after 5 h and subsided after 6 h but remained elevated relative to untreated cells. These findings indicate that PGE2 increases the occupancy of endogenous C/EBP-β on IL-8. As C/EBP-β and p300 are thought to assemble on promoters, thus forming a transcription complex,Citation16-Citation18 we verified whether the PGE2-induced C/EBP-β recruitment regulates also the appearance of p300 on IL-8 promoter and activates its transcription. A Re-ChIP assay was performed to reach this aim. The second IP with anti-p300 identifies p300 in the C/EBP-β immunoprecipitate, indicating that a complex containing both p300 and C/EBP-β occupies the same IL-8 promoter DNA. Conversely, when the first anti-p300 immunoprecipitate was similarly re-immunoprecipitated with anti-C/EBP-β, C/EBP-β was also identified in the p300 complex associated with IL-8 DNA. PGE2 also caused marked enhancement of phosphorylated RNA polymerase II (P-RNAP II) recruitment to the IL-8 gene promoter. It is worth noting that the timing for association of C/EBP-β and p300 with the promoter and for activation of RNAP II paralleled the timing of activation of IL-8 transcription that we have previously observed.Citation14
Figure 5. PGE2 increases occupancy of endogenous C/EBP-β and p300 on the IL-8 promoter and H3 acetylation. (A) 1321N1 cells were treated with PGE2 for the indicated times. Primary ChIP was performed using anti-C/EBP-β, anti-p300, anti-P-RNAP II, anti-Ac-H3 (K9), anti-Ac-H3 (K14), and anti-Ac-H4. The beads from the first IP were washed and eluted for the second IP. The eluate from the first IP with anti-C/EBP-β was used for the second IP with anti-p300 or no antibody as control (2nd IP, top). The eluate from the first IP with anti-p300 was used for the second IP with anti-C/EBP-β or no antibody as control (2nd IP, bottom). Brackets indicate which of the first IP was used for second IP. Similar data were obtained in another experiment. RT–PCR (A) and Real-time PCR analysis (B) of ChIPs were performed as described in Materials and Methods.
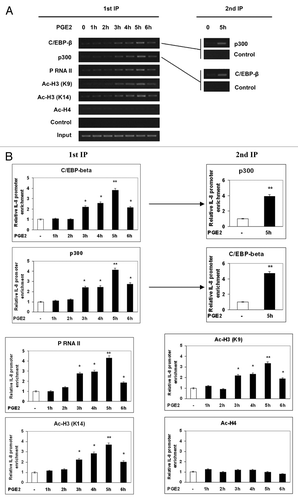
We then investigated whether p300 recruitment via association with C/EBP-β works in coordination with histone acetylation during PGE2-induced derepression of the IL-8 gene. As shown in , association of the IL-8 gene promoter with acetylated histone H3 at K9 or K14 was significantly increased in the presence of PGE2. Similar results were observed in the A172 and NHA cell lines (Fig. S2).
These findings indicate that PGE2 induces the recruitment of p300 and C/EBP-β at the IL-8 promoter as well as histone H3 hyperacetylation.
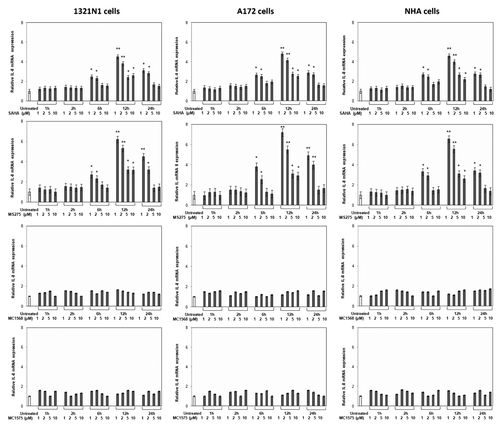
Class I HDAC inhibitors increase IL-8 gene transcription
In order to verify whether IL-8 gene expression was regulated by histone acetylation state, we treated cells with SAHA (a non-selective HDACi), MS275 (a class I-selective HDACi), MC1568 (a class II-selective HDACi) and MC1575 (a class II-selective HDACi), and measured IL-8 mRNA and protein levels. The results, shown in , indicated that both SAHA and MS275 induced a concentration-dependent increase of IL-8 mRNA levels, starting 6 h after treatment in each cell type. Conversely, no effects were observed when cells were treated with MC1568 and MC1575. The mRNA levels of IL-8 were consistent with protein content (data not shown).
Figure 6. Effects of HDACis on IL-8 expression. 1321N1, A172 and NHA cells were treated with the indicated concentrations of drugs for the indicated times and total RNA was analyzed by real-time PCR. Data points represent the average of triplicate determinations ± SD. Statistical analyses were performed compared with respective untreated control cells. *p < 0.01; **p < 0.001; absence of asterisks, not significant.
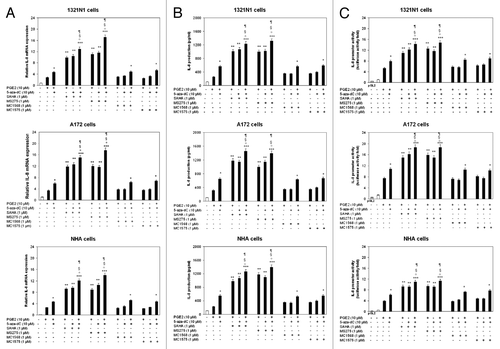
HDACis act with 5-aza-dC to increase PGE2-induced IL-8 gene transcription
Since both HDACis and 5-aza-dC could synergistically activate the transcription of important genes in cancer cells,Citation19 we examined the effects of HDACis, either alone or in concert with 5-aza-Dc, on PGE2-dependent IL-8 expression. We showed that SAHA and MS275 induce a drastic enhancement of PGE2-mediated IL-8 expression at mRNA (), protein () and promoter () level (p < 0.01). The combination of SAHA or MS275 with 5-aza-dC resulted in a much more significant IL-8 increase (p < 0.001).
Figure 7. Combined effects of PGE2, 5-aza-dC and HDACis on IL-8 expression. 1321N1, A172 and NHA cells were treated with 5-aza-dC (10 μM for 72 h), HDAC inhibitors (1μM) for 12 h, and PGE2 (10 μM) for 5 h, alone or in combination. (A) Total RNA was extracted and subjected to real-Time PCR analysis. IL-8 mRNA levels were normalized to β-actin levels and expressed as relative to untreated control cells. (B) The amount of IL-8 protein was measured by ELISA. (C) Cells were transiently transfected with 1 µg of the IL-8 promoter construct and subsequently treated as above mentioned. Data are expressed as means ± SD of results in three independent experiments. Significance: *p < 0.05, **p < 0.01, and ***p < 0.001, as compared with PGE2-treated cells; °p < 0.001, as compared with PGE2/5-aza-dC-treated cells; §p < 0.01, as compared with PGE2/HDACi -treated cells; ¶p < 0.01, as compared with 5-aza-dC/HDACi -treated cells.
Effect of PGE2, 5-aza-dC and HDACis on the methylation status of CpG site 5 and DNMTs/HDACs recruitment
The methylation status of CpG site 5 on the IL-8 promoter was studied by MeDIP analysis in 1321N1 astrocytoma cells untreated or treated with PGE2, 5-aza-dC, SAHA and MS275, alone or in combination. clearly shows that treatment with PGE2 or 5-aza-dC alone led to a significant reduction in anti-mecyt antibody binding to the IL-8 promoter region containing CpG site 5. This reduction, induced by PGE2, started 1 h post-treatment, with the greatest inhibition observed after 5 h. Combination of PGE2 and 5-aza-dC further reduced the methylation status at CpG site 5 when compared with each single treatment, whereas the combination of PGE2 with either SAHA or MS275 did not affect the pattern of methylation modified by PGE2 alone. We then determined the effects of PGE2 alone or in combination with 5-aza-dC, SAHA and M2752 on the association of the major DNMTs identified in human, namely DNMT1, DNMT3a and DNMT3b, with the IL-8 promoter, using ChIP assays. As shown in , treatment with PGE2 reduced the level of association of DNMT1 and DNMT3a with the IL-8 promoter, with a maximum effect occurring 5 and 1 h after treatment, respectively. Co-treatment with 5-aza-dC led to synergistic dissociation of DNMT1 from the IL-8 promoter, whereas combined treatment with PGE2 and SAHA or MS275 resulted in a profile similar to that observed with PGE2 alone. In striking contrast, under all treatments, there was little association of DNMT3b with the IL-8 promoter.
Figure 8. CpG site 5 methylation and DNMTs/HDACs recruitment to this area in cells treated with PGE2, 5-aza-dC and HDACis, alone or in combination. 1321N1 cells were treated with PGE2 (10 μM) from 1 through 5 h. For combination experiments with 5-aza-dC (10 μM for 72 h) and HDAC inhibitors SAHA and MS275 (1μM for 12 h), PGE2 (10 μM) was used at the most effective times on CpG site 5 demethylation and DNMT/HDAC association to IL-8 promoter. (A) MeDIP with anti-methyl-cytosine antibody and semiquantitative PCR analysis were performed as described in Materials and Methods. Primers covering the CpG site 5 in IL-8 promoter region were used. (B) The immunoprecipitated complexes were tested by real time-PCR as described in Materials and Methods. (C) ChIP (chromatin immunoprecipitation) with anti DNMT1, DNMT3a, DNMT3b, HDAC1, HDAC2 and HDAC3 antibodies followed by semiquantitative PCR analysis was conducted using primers as in A. (D) The immunoprecipitated complexes were tested by real time-PCR as described in Materials and Methods. Data are expressed as means ± SD of results in three independent experiments.Significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, as compared with untreated cells.
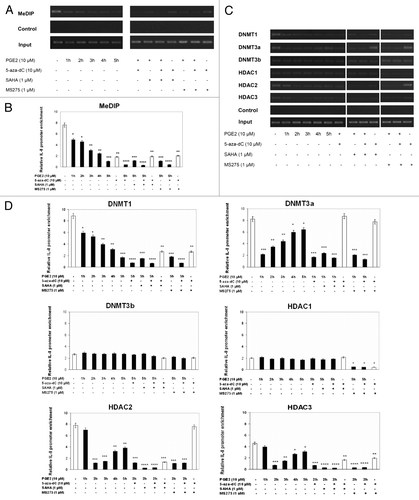
Given the cooperative linkage existing between DNMTs and HDACs in the epigenetic control of gene expression, we determined the possible association of HDACs with the IL-8 promoter in the presence of PGE2, alone or in combination with 5-aza-dC, SAHA and MS275. We focused on the main HDACs involved in H3 histone acetylation, including HDAC1, HDAC2 and HDAC3,Citation20-Citation22 and performed immunoprecipitation experiments with specific antibodies (). Dissociation and or masking of HDAC2 and 3 was highly significant 2 h after PGE2 treatment. A more significant decrease was observed following co-exposure to PGE2/SAHA, whereas dual treatment with PGE2 and MS275 resulted in an additive reducing effect only on HDAC3 recruitment. Combination treatments with PGE2/5-aza-dC and SAHA/5-aza-dC had similar effects as single treatment with PGE2. The association of HDAC1 with the IL-8 promoter was not modified by PGE2, used either alone or in combination with SAHA and 5-aza-dC. On the contrary, co-exposure to PGE2/MS275 or 5-aza-dC/MS275 significantly reduced HDAC1 recruitment.
Discussion
Over the last decade the contribution of epigenetic alterations to the tumorigenic process has been studied worldwide, generating promising results. Patterns and levels of DNA methylation and histone acetylation are the most studied epigenetic modifications in the context of gene transcription and abnormal events that lead to oncogenic process. Evidence suggests that these marks are dynamically linked in the epigenetic control of gene expression and that their dysregulation plays an important role in tumorigenesis. In the present study we were interested in investigating the role of PGE2 as a potential epigenetic inducer of IL-8 gene in human astrocytoma, given its increased secretionCitation23 and activating effects on IL-8 expressionCitation14,Citation24 in this type of tumor. Various aspects of the IL-8 gene regulation in response to inflammatory stimuli or in neoplastic conditions have been extensively studied.Citation25-Citation28 However, in contrast to the elucidation of the cis element and trans factors involved in the transcriptional control of this gene, the evidence for the participation of epigenetic mechanisms was only partially revealed, and contrasting results were sometimes obtained. In this regard, a marked CpG hypomethylation of the IL-8 promoter has been detected in human colorectal adenocarcinomasCitation29 and in aggressive and chronic periodontitis,Citation30,Citation31 whereas other studies have proved that atypical methylation pattern of the IL-8 promoter strongly correlates with an increase in IL-8 expression and the metastatic potential of breast carcinoma cells.Citation32 A larger number of studies have been conducted to determine histone modifications associated with IL-8 gene expression following a variety of stimuli, such as LPS in intestinal epithelial cells,Citation33 leptin in synovial fibroblast cells,Citation34 UVR in keratinocytesCitation35 and Moraxella catarrhalis in bronchial epithelial cells.Citation36 On the basis of these findings, here we have explored potential epigenetic cross-talk triggered by PGE2 responsible for the aberrant expression of IL-8 in human astrocytoma. Our results provided compelling evidence to indicate that PGE2 activates IL-8 transcription through: (1) specific demethylation of the CpG site 5 located within the C/EBP-β consensus sequence in the IL-8 promoter, as supported by bisulfite sequencing () and MeDIP analysis () and (2) abnormal acetylation of histone H3 in this region (). Our results showed that PGE2 or 5-aza-dC alone, and much more in combination with each other, demethylated CpG site 5 and reduced DNMT1 and DNMT3a association with this region. The reduction was already significant 1 h after PGE2 treatment, but the greatest dissociation occurred earlier for DNMT3a (1 h) than for DNMT1 (5 h) (). Although several reports provide evidence that PGE2 increases expression of DNMT1 and DNMT3a in many cell types and a wide variety of diseases,Citation37,Citation38 the present findings are consistent with recent data reporting that PGE2 decreased the expression of DNMT1 and DNMT3a in RAW macrophages.Citation38 It can therefore be assumed that PGE2 effects on DNA methylation machinery are cell type-specific. Our results are indicative of the role of PGE2 in inducing DNA demethylation, as they refer to the levels of interaction of DNMTs to the IL-8 promoter. However, we elucidated a more complex epigenetic role of PGE2 on IL-8 expression, as we showed that HDAC2 and HDAC3 association at the IL-8 promoter was significantly reduced at 2 h after PGE2 treatment (). These effects were enhanced by SAHA co-treatment, whereas co-exposure to MS275 enhanced only the effect of PGE2 on HDAC3. ChIP experiments showing that PGE2 or SAHA reduced the recruitment of HDAC2 and 3 to the IL-8 promoter and that MS275 affected the presence of HDAC1 and 3 suggest that inhibition of HDAC3 is sufficient and necessary to activate IL-8 promoter. The kinetics of DNA demethylation and histone acetylation induced by PGE2 clearly indicates that demethylation at CpG site 5 region preceded and perhaps favored the successive chromatin hyperacetylation. These results imply that DNA demethylation could trigger events needed for higher levels of histone acetylation. The findings shown here support a model of chromatin structure changes in human astrocytoma where PGE2 orchestrates a coordinate process of specific CpG demethylation followed by histone H3 hyperacetylation which lead to IL-8 transcription through recruitment of C/EBP-β and p300 at IL-8 gene regulatory region. This previously unrecognized mechanism is supported by the significant role of CpG site 5 demethylation—induced by PGE2—on IL-8 overexpression, as shown by either a promoter targeted siRNA method () or a transient reporter assay using a reporter vector in which only IL-8 promoter CpG site 5 of is methylated (). Identifying specific methylated sites is important for understanding the mechanisms behind regulating gene expression, as it is known that the interaction between only one CpG site and DNA enhancer elements can be sufficient to modify gene transcription.Citation39 A strong regulation of the IL-8 gene by HDAC inhibitors has been reported in several cell systems.Citation40 To this regard, it is worth mentioning that, in most instances, HDAC inhibitors were positively acting mainly in cooperation with several inducers of IL-8 gene expression, such as IL-1βCitation41-Citation43 or TNF-α.Citation44-Citation46 On the contrary, in some cell types, IL-8 expression was downregulated by HDAC inhibitors.Citation47 Here we showed that in astrocytoma cells SAHA and MS275 acted either alone () or in synergy with PGE2 or 5-aza-dC () in inducing chromatin remodeling and enhancing IL-8 transcription. The fact that the effects of DNA demethylating and histone hyperacetylating treatments reinforce each other confirms that these events occur in sequence in the same pathway that leads to activation of IL-8 gene. Indeed, a strong link between lineage-specific CpG demethylation events and histone modifications was recently suggested.Citation33,Citation48-Citation50 Aberrant DNA methylation/demethylation and histone deacetylation/hyperacetylation are highly regulated and dynamic processes that may coexist in cancer cells, altering gene expression in opposite directions. No previous data were available suggesting a potential combinatory effect of PGE2 on both DNA promoter methylation and histone acetylation, as we showed here. The role of PGE2 on specific CpG site methylation/demethylation status has not been investigated yet in any cell system, and its involvement in histone acetylation has been shown only in few cases. In breast cancer cells, PGE2 induced histone H3 hyperacetylation and increased the binding of RNAP II to the liver receptor homolog-1 (LRH-1) promoter.Citation51 In mink uterine stromal cells, PGE2 induced an increase in histone H3 acetylation, inducing the transactivation of steroidogenic acute regulatory protein (StAR)Citation52 and vascular endothelial growth factor (VEGF)Citation53 genes.
In summary, our results support the existence of an epigenetic interplay that marks the PGE2-mediated IL-8 activation in human astrocytoma. Results reported here provide a novel gene derepression mechanism, emerging from integrated changes in the levels of cytosine methylation and histone acetylation, which control IL-8 transcription. Based on previous results showing that IL-8 can account for the higher aggressiveness of astrocytoma cells,Citation54-Citation56 our data suggest that: (1) pharmacological reversal of hypomethylation could be used as a useful tool for blocking astrocytoma progression into the aggressive and metastatic stages of the disease and (2) the use of HDAC inhibitors in anti-glioma strategies, in particular those with class I-specific activity, could have adverse effects by increasing the expression of pro-inflammatory molecules.
Materials and Methods
Reagents
Prostaglandin E2 (PGE2), 5-aza-2-deoxyazacytidine (5-aza-dC), SAHA and MS-275 were purchased from Sigma (Sigma-Aldrich). MC1575 and MC1568 were synthesized as previously described.Citation57,Citation58 Restriction endonucleases, S-adenosyl-l-methionine (AdoMet), T4 DNA Ligase, DNA polymerases, DNA size standards and competent cells were from New England Biolabs (Ipswich, MA). Recombinant human C/EBP-β was purchased from Novusbio.
Tissue samples
Forty-six freshly resected astrocytic tumor specimens were collected during the surgeries at the Department of Neurosciences of the University Hospital of Messina from October 2008 to December 2011. The tumors were located in the cerebral hemisphere (10), in the cerebellum (21), in the optic pathway/hypothalamus (4), in the thalamus (5) and in the brainstem (6). In 14 cases the tumors were diagnosed as low-grade astrocytoma, in 13 cases as anaplastic astrocytoma, and in 19 cases as glioblastoma. Patient data are shown in . The histological grade of these tumors was classified according to the World Health Organization (WHO) criteria. Soon after any surgical removal, the specimens were frozen in liquid nitrogen and stored at -80°C until any further use. This investigation adhered to the Declaration of Helsinki and was approved by the Ethics Committee of the University Hospital of Messina. An informed consent was given by the patients.
Table 1. Patient characteristics
Cells and culture conditions
Normal human astrocytes (NHA) were purchased from Lonza and they were cultured in the provided astrocyte growth media supplemented with rhEGF, insulin, ascorbic acid, GA-1000, L-glutamine and 5% FBS. Human 1321N1 (derived from grade II astrocytoma) and A-172 (derived from grade IV glioblastoma) cell lines (American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, Milano, Italy) supplemented with 100 units/ml penicillin (EuroClone), 100 μg/ml streptomycin (EuroClone), and 10% (v/v) fetal bovine serum (FBS; Sigma-Aldrich). Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C.
Demethylation with the DNA demethylating agent 5-aza-dC
Cells were treated with DNA demethylating agent 5-aza-dC by addition of fresh medium containing 5-aza-dC (10 μmol/L) every day for 3 consecutive days. Cells were harvested for DNA and RNA extractions.
DNA extraction
The isolation of DNA from fresh/frozen tissue specimens was performed by QIAamp DNA Mini kit (Qiagen Inc.). Total genomic DNA was extracted by cells using the TRIZOL reagent (Invitrogen, Carlsbad) according to the manufacture’s recommendation.
Bisulphite sequencing
Five hundred ng of DNA was treated with sodium bisulphite using the Epitect Bisulfite kit (Qiagen). One microliter of bisulphite-modified DNA containing ~10 ng DNA (range ~4–20 ng) was amplified using primers common to the methylated and unmethylated DNA sequences. Cell PCR products were cloned into the pGEM-T Easy vector (Promega), and 20 clones from each sample were sequenced in forward and reverse directions with the CEQ DTCS Quick Start Kit (Beckman Coulter S.p.A.), and with an automated DNA sequencer (Beckman Coulter CEQ 2000 Analysis System). The oligonucleotide primers used were as follows:
sites 1–2 forward, 5′-AAGTTTTAGGTTGGTTGGAGAAAGA-3′
sites 1–2 reverse, 5′-CCTCTAAAAACCCATAATCATCAAC-3′
sites 3–6 forward, 5′-GTTATATGGTTTATGATAAAGTTATTTAG-3′
sites 3–6 reverse, 5′-CCAATAATTTCTTCCTAACTCTTATC-3′.
RNA Extraction and Reverse Transcription
Total RNA was extracted with TRIZOL (Invitrogen) from resected tissues and cells. One µg of total RNA was reverse-transcribed in a total volume of 20 µl with IMProm-IITM reverse transcriptase kit (Promega) by using oligo-dT primers, according to the manufacturer’s instructions.
Quantitative Real-Time PCR
Quantitative Real-Time PCR (qRT-PCR) was performed using the ABI Prism 7500 Real-Time PCR System (Applied Biosystems). The sequences of primers and probe used for specific IL-8 PCR amplification were as follows: 5′-CTCACTGTGTGTAAACATGACTTCCA-3′ (primer forward), 5′-TTCACACAGAGCTGCAGAAATCA-3′ (primer reverse) and 5′-FAM-CCGTGGCTCTCTTTGGCAGCCTTC-MGBNFQ-3′ (probe). Thermal cycling conditions included activation at 95°C (10 min) followed by 40 cycles each of denaturation at 95°C (15 sec) and annealing/elongation (1 min) at 60°C. Each sample was analyzed in triplicate with β actin (Applied Biosystems) as housekeeping gene, and the mean values of IL-8 were calculated. The cycle threshold (Ct) was used to calculate relative amounts of target DNA. The Ct was determined as the number of PCR cycles required for a given reaction to reach an arbitrary fluorescence value within the linear amplification range.
Measurement of IL-8 levels
Cell-free culture supernatants and the extracts from biopsy specimens were analyzed for IL-8 protein content using a commercial enzyme immunoassay kit according to the manufacturer’s instructions (Bio Source International). Measurement was done in triplicate. The values of IL-8 protein were normalized to the total protein values of each sample.
Luciferase reporter assay using a promoter with specific DNA methylation of CpG site 5
The 5′ region of human IL-8 was amplified using the following primers containing artificial KpnI and XhoI restriction sites: (1) sense, 5′-ggtacCTCAAGTCTTAGGTTGGTTG-3′and (2) antisense, 5′-ctcgaGCTCTGCTGTCTCTGAAAGT-3′. The resulting 1497-bp PCR product was subsequently cloned into the pCR 2.1-TOPO vector using the TOPO TA cloning kit (InVitrogen). The pCR 2.1 TOPO plasmid containing the IL-8 promoter insert (hereafter referred to as TOPO-IL-8) was linearized with KpnI and methylated in vitro using SssI methylase, which non-specifically methylates all CpG dinucleotides. Resistance to cleavage by the methylation-sensitive restriction enzyme MspJI confirmed the efficiency of in vitro methylation. The linearized methylated and unmethylated TOPO-IL-8 vectors were then digested with XhoI to excise the IL-8 promoter fragments. After fractionation on a 1% agarose gel, the DNA bands corresponding to 1497 bp were cut from the gel, isolated using GenElute Spin columns (Supelco), ethanol-precipitated, and subcloned as a KpnI/XhoI fragment into the pGL3-Basic vector (Promega) (hereafter referred to as pGL3-IL-8-meth and pGL3-IL-8-unmeth).
It was constructed a reporter vector in which only this site of IL-8 promoter was methylated in order to analyze the effect of CpG site 5 methylation on IL-8 gene expression. The region encompassing the CpG residue 5 was generated by PCR amplification with the following primer set: (1) sense, 5′-TTTGATAAGGAACAAATAGGAAGTG-3′ and (2) antisense, 5′-TAGGGTGATGATATAAAAAGCCAC-3′ and inserted into pCR 2.1-TOPO vector using the TOPO TA cloning kit (Invitrogen). The pCR 2.1 TOPO plasmid containing the desired region of IL-8 promoter (hereafter referred to as TOPO- IL-8 CpG site 5 region) was linearized with KpnI and methylated in vitro using SssI methylase. The efficiency of in vitro methylation was confirmed by resistance to cleavage by the methylation-sensitive restriction enzyme MspJI. The linearized methylated and unmethylated TOPO-IL-8 CpG site 5 region vectors were then digested with XhoI. After fractionation on a 1% agarose gel, the DNA bands corresponding to 236 bps were cut from the gel, isolated using GenElute Spin columns (Supelco), and ethanol-precipitated. The fragments were then ligated into pGL3-Basic vector (Promega) between the KpnI and XhoI restriction sites (hereafter referred to as pGL3-IL-8 CpG site 5 region-meth and pGL3-IL-8 CpG site 5 region-unmeth).
Transient transfections
Cells were plated in 35-mm well plates (5 × 106 cells/well) in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, Milano, Italy) without fetal bovine serum and transiently transfected with 1 µg of IL-8 constructs using FuGENE_6 Transfection Reagent (Roche Diagnostics) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were treated with 5-aza-dC (10 μΜ for 72 h), HDAC inhibitors (at various concentrations and times), and PGE2, alone or in combination. Consistent with our previous data,Citation14 5-h treatment and 10 µM PGE2 were used. Then, cells were harvested and protein extracts were prepared for the luciferase activity using luciferine (Promega) as the substrate. In each transfection, 0.2 µg of pSVβgal was added and the luciferase activity was normalized with respect to the β-galactosidase activity.
RNAi analysis
We used the pcPUR hU6 vector (iGENETherapeutics, Inc.; www.iGENE-therapeutics.co.jp), which contains the human U6 promoter, a puromycin resistance gene, and BspMI cloning sites, to construct the siRNA expression vector. The sequences of the siRNAs used were 5′-GGCCATCAGTTGCAAATCGTG-3′ (IL8 promoter) and 5′-GGCCATCAGTTGCAAATAATG-3′ (control).
We synthesized oligonucleotides with hairpin, terminator, and overhanging sequences. We annealed these fragments and inserted them into the BspMI sites of pcPUR hU6. These siRNA constructs were transfected into astrocytoma cells using GeneJuice (Novagen). Transfected cells were selected in medium supplemented with puromycin (2.5 μg/ml) for 2–4 d. Vector-transfected cells were used as control. Live cells were isolated on a Ficoll gradient and used for analyses.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts, prepared with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce), were subjected to electrophoretic mobility shift assay using the LightShift Chemiluminescent kit (Pierce). The binding reaction mixture containing 3 µg of nuclear extract or 20 ng recombinant human C/EBP-β protein, 20 fmol of 5′ biotin-labeled oligonucleotide probe(s), 1× binding buffer, 50 ng of poly(dI_dC), 2.5% glycerol, 0.05% Nonidet P-40, and 5 mM MgCl2 was incubated at room temperature for 20 min in a final volume of 20µl. For supershift assays, nuclear extracts were incubated with 2 µg of anti-C/EBP-β antibody (Santa Cruz Biotechnology) at room temperature (for 20 min) prior to probe addition. An unmethylated (UnM) or methylated (M) probe (C/EBP-β probe) encompassing the C/EBP-β binding site and containing a mutation of the adjacent NF-κB consensus sequence was used: 5′-TCAGTTGCAAATCGTGttcgggttaCTCTGACATAATGAAAAGAT-3′. For competition assays, a 200-fold excess of unlabeled double-stranded oligonucleotides was incubated with the extracts at room temperature for 10 min before probe addition. Bound complexes were separated on 8% polyacrylamide gels, blotted onto membrane, and visualized by autoradiography (Hyperfilm, Fuji).
Chromatin Immunoprecipitation (ChIP) Assay
The ChIP enzymatic assay (Active Motif) was performed and the sheared chromatin samples were used for immunoprecipitation with 2 µg of anti-C/EBP-β (Santa Cruz Biotechnology), anti-p300 (Santa Cruz Biotechnology), anti-phosphorylated RNAP II (P-Pol II) (Covance Research Products), anti-Ac-H3 (K9) (Upstate Biotechnology), anti-Ac-H3 (K14) (Upstate Biotechnology), anti-Ac-H4 (Upstate Biotechnology), anti-methylcytosine (anti-mecyt; Anaspec, Fremont, CA), anti-DNMT1 (Abcam), anti-DNMT3a (Abcam), anti-DNMT3b (Abcam), anti-HDAC1 (Abcam), anti-HDAC2 (Abcam) and anti-HDAC3 (Abcam) antibodies overnight at 4°C. Immunocomplexes were subjected to cross-link reversal, extracted, and precipitated as described in the protocol. The eluted DNA and the aliquots of chromatin prior to immunoprecipitation (input) were amplified by PCR. To detect the DNA sequence from position -150 to +11 of the IL-8 gene promoter, in which the CpG site 5 is located, we used the following primer set: forward, 5′-GATAAGGAACAAATAGGAAGTGTG-3′; reverse, 5′-TGGCTTTTTATATCATCACCCTAC-3′.
The PCR conditions were as follows: 95°C for 4 min and 38 cycles of 94°C for 50 sec, 65°C for 50 sec, and 72°C for 1 min. PCR products were separated by 2% agarose gel containing ethidium bromide.
Real-time PCR quantification analysis of immunoprecipitated DNA
Applied Biosystems 7500 system SDS software was used for real-time PCR analysis of ChIP experiments. The analysis was performed using SYBR Green DNA Master mix (Applied Biosystems). Absolute quantification was performed using serial dilutions of the input DNA samples to obtain standard curves. The values of negative controls (no antibody) were subtracted from the corresponding samples. Immunoprecipitated fractions were estimated by human β-actin housekeeping gene amplification. Oligonucleotide sequences used for real-time PCR are available upon request.
Statistical analysis
The one-way analysis of variance (ANOVA) test, followed by a pair-wise multiple comparison test (Bonferroni t test), was performed to identify the differences among the groups. The Pearson’s correlation test was employed to analyze the correlation between CpG methylation status and IL-8 expression levels. Differences were considered significant when the p value (two-sided) was < 0.05. R software (version 2.10.0; www.r-project.org) was used for performing statistical analysis.
Additional material
Download Zip (368.4 KB)Acknowledgments
The authors gratefully acknowledge Dr Germana Cubeta for her assistance in the linguistic revision of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Disclosures
This work was supported by grants for Project of National Relevance (PRIN) 2008 “Study on the effects of new histone deacetylase inhibitors on cell physiology and pathology: implications for cancer and metabolic diseases and their treatment” coordinated by Prof. Diana Teti.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/22446
Related Research Data
References
- Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science 1999; 286:481 - 6; http://dx.doi.org/10.1126/science.286.5439.481; PMID: 10521337
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074 - 80; http://dx.doi.org/10.1126/science.1063127; PMID: 11498575
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429:457 - 63; http://dx.doi.org/10.1038/nature02625; PMID: 15164071
- Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 2012; 22:50 - 5; http://dx.doi.org/10.1016/j.gde.2012.02.008; PMID: 22402447
- Lambert MP, Herceg Z. Epigenetics and cancer, 2nd IARC meeting, Lyon, France, 6 and 7 December 2007. Mol Oncol 2008; 2:33 - 40; http://dx.doi.org/10.1016/j.molonc.2008.03.005; PMID: 19383327
- Park YJ, Claus R, Weichenhan D, Plass C. Genome-wide epigenetic modifications in cancer. Prog Drug Res 2011; 67:25 - 49; PMID: 21141723
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 2006; 31:89 - 97; http://dx.doi.org/10.1016/j.tibs.2005.12.008; PMID: 16403636
- Vaissière T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 2008; 659:40 - 8; http://dx.doi.org/10.1016/j.mrrev.2008.02.004; PMID: 18407786
- Lin Y, Huang R, Chen L, Li S, Shi Q, Jordan C, et al. Identification of interleukin-8 as estrogen receptor-regulated factor involved in breast cancer invasion and angiogenesis by protein arrays. Int J Cancer 2004; 109:507 - 15; http://dx.doi.org/10.1002/ijc.11724; PMID: 14991571
- Yao C, Lin Y, Chua MS, Ye CS, Bi J, Li W, et al. Interleukin-8 modulates growth and invasiveness of estrogen receptor-negative breast cancer cells. Int J Cancer 2007; 121:1949 - 57; http://dx.doi.org/10.1002/ijc.22930; PMID: 17621625
- Wang XB, Tian XY, Li Y, Li B, Li Z. Elevated expression of macrophage migration inhibitory factor correlates with tumor recurrence and poor prognosis of patients with gliomas. J Neurooncol 2012; 106:43 - 51; http://dx.doi.org/10.1007/s11060-011-0640-3; PMID: 21725855
- Piperi C, Samaras V, Levidou G, Kavantzas N, Boviatsis E, Petraki K, et al. Prognostic significance of IL-8-STAT-3 pathway in astrocytomas: correlation with IL-6, VEGF and microvessel morphometry. Cytokine 2011; 55:387 - 95; http://dx.doi.org/10.1016/j.cyto.2011.05.012; PMID: 21684758
- Bonavia R, Inda MM, Vandenberg S, Cheng SY, Nagane M, Hadwiger P, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene 2011; 31:4054 - 66; http://dx.doi.org/10.1038/onc.2011.563; PMID: 22139077
- Venza M, Visalli M, Alafaci C, Caffo M, Caruso G, Salpietro FM, et al. Interleukin-8 overexpression in astrocytomas is induced by prostaglandin E2 and is associated with the transcription factors CCAAT/enhancer-binding protein-β and CCAAT/enhancer-binding homologous protein. Neurosurgery 2011; 69:713 - 21, discussion 721; http://dx.doi.org/10.1227/NEU.0b013e31821954c6; PMID: 21471847
- Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004; 305:1289 - 92; http://dx.doi.org/10.1126/science.1101372; PMID: 15297624
- Mink S, Haenig B, Klempnauer KH. Interaction and functional collaboration of p300 and C/EBPbeta. Mol Cell Biol 1997; 17:6609 - 17; PMID: 9343424
- Kovács KA, Steinmann M, Magistretti PJ, Halfon O, Cardinaux JR. CCAAT/enhancer-binding protein family members recruit the coactivator CREB-binding protein and trigger its phosphorylation. J Biol Chem 2003; 278:36959 - 65; http://dx.doi.org/10.1074/jbc.M303147200; PMID: 12857754
- Cui TX, Piwien-Pilipuk G, Huo JS, Kaplani J, Kwok R, Schwartz J. Endogenous CCAAT/enhancer binding protein beta and p300 are both regulated by growth hormone to mediate transcriptional activation. Mol Endocrinol 2005; 19:2175 - 86; http://dx.doi.org/10.1210/me.2004-0502; PMID: 15860545
- Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 2007; 74:659 - 71; http://dx.doi.org/10.1016/j.bcp.2007.04.007; PMID: 17498667
- Li X, Kaplun A, Lonardo F, Heath E, Sarkar FH, Irish J, et al. HDAC1 inhibition by maspin abrogates epigenetic silencing of glutathione S-transferase pi in prostate carcinoma cells. Mol Cancer Res 2011; 9:733 - 45; http://dx.doi.org/10.1158/1541-7786.MCR-10-0505; PMID: 21622623
- Jin JS, Tsao TY, Sun PC, Yu CP, Tzao C. SAHA inhibits the growth of colon tumors by decreasing histone deacetylase and the expression of cyclin D1 and survivin. Pathol Oncol Res 2012; 18:713 - 20; http://dx.doi.org/10.1007/s12253-012-9499-7; PMID: 22270866
- Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J 2007; 26:412 - 23; http://dx.doi.org/10.1038/sj.emboj.7601516; PMID: 17245431
- Payner T, Leaver HA, Knapp B, Whittle IR, Trifan OC, Miller S, et al. Microsomal prostaglandin E synthase-1 regulates human glioma cell growth via prostaglandin E(2)-dependent activation of type II protein kinase A. Mol Cancer Ther 2006; 5:1817 - 26; http://dx.doi.org/10.1158/1535-7163.MCT-05-0548; PMID: 16891468
- Samaras V, Piperi C, Levidou G, Zisakis A, Kavantzas N, Themistocleous MS, et al. Analysis of interleukin (IL)-8 expression in human astrocytomas: associations with IL-6, cyclooxygenase-2, vascular endothelial growth factor, and microvessel morphometry. Hum Immunol 2009; 70:391 - 7; http://dx.doi.org/10.1016/j.humimm.2009.03.011; PMID: 19332096
- Strieter RM, Belperio JA, Burdick MD, Sharma S, Dubinett SM, Keane MP. CXC chemokines: angiogenesis, immunoangiostasis, and metastases in lung cancer. Ann N Y Acad Sci 2004; 1028:351 - 60; http://dx.doi.org/10.1196/annals.1322.041; PMID: 15650260
- Romagnani P, Lasagni L, Annunziato F, Serio M, Romagnani S. CXC chemokines: the regulatory link between inflammation and angiogenesis. Trends Immunol 2004; 25:201 - 9; http://dx.doi.org/10.1016/j.it.2004.02.006; PMID: 15039047
- Mukaida N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol 2003; 284:L566 - 77; PMID: 12618418
- Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res 2008; 14:6735 - 41; http://dx.doi.org/10.1158/1078-0432.CCR-07-4843; PMID: 18980965
- Dimberg J, Ström K, Löfgren S, Zar N, Lindh M, Matussek A. DNA promoter methylation status and protein expression of interleukin-8 in human colorectal adenocarcinomas. Int J Colorectal Dis 2011; 27:709 - 14; PMID: 22108905
- Andia DC, de Oliveira NF, Casarin RC, Casati MZ, Line SR, de Souza AP. DNA methylation status of the IL8 gene promoter in aggressive periodontitis. J Periodontol 2010; 81:1336 - 41; http://dx.doi.org/10.1902/jop.2010.100082; PMID: 20450371
- Oliveira NF, Damm GR, Andia DC, Salmon C, Nociti FH Jr., Line SR, et al. DNA methylation status of the IL8 gene promoter in oral cells of smokers and non-smokers with chronic periodontitis. J Clin Periodontol 2009; 36:719 - 25; http://dx.doi.org/10.1111/j.1600-051X.2009.01446.x; PMID: 19659670
- De Larco JE, Wuertz BR, Yee D, Rickert BL, Furcht LT. Atypical methylation of the interleukin-8 gene correlates strongly with the metastatic potential of breast carcinoma cells. Proc Natl Acad Sci U S A 2003; 100:13988 - 93; http://dx.doi.org/10.1073/pnas.2335921100; PMID: 14623984
- Angrisano T, Pero R, Peluso S, Keller S, Sacchetti S, Bruni CB, et al. LPS-induced IL-8 activation in human intestinal epithelial cells is accompanied by specific histone H3 acetylation and methylation changes. BMC Microbiol 2010; 10:172; http://dx.doi.org/10.1186/1471-2180-10-172; PMID: 20546607
- Tong KM, Shieh DC, Chen CP, Tzeng CY, Wang SP, Huang KC, et al. Leptin induces IL-8 expression via leptin receptor, IRS-1, PI3K, Akt cascade and promotion of NF-kappaB/p300 binding in human synovial fibroblasts. Cell Signal 2008; 20:1478 - 88; http://dx.doi.org/10.1016/j.cellsig.2008.04.003; PMID: 18501560
- Pollack BP, Sapkota B, Boss JM. Ultraviolet radiation-induced transcription is associated with gene-specific histone acetylation. Photochem Photobiol 2009; 85:652 - 62; http://dx.doi.org/10.1111/j.1751-1097.2008.00485.x; PMID: 19076306
- Slevogt H, Schmeck B, Jonatat C, Zahlten J, Beermann W, van Laak V, et al. Moraxella catarrhalis induces inflammatory response of bronchial epithelial cells via MAPK and NF-kappaB activation and histone deacetylase activity reduction. Am J Physiol Lung Cell Mol Physiol 2006; 290:L818 - 26; http://dx.doi.org/10.1152/ajplung.00428.2005; PMID: 16399788
- Xia D, Wang D, Kim SH, Katoh H, DuBois RN. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat Med 2012; 18:224 - 6; http://dx.doi.org/10.1038/nm.2608; PMID: 22270723
- Huang SK, Scruggs AM, Donaghy J, McEachin RC, Fisher AS, Richardson BC, et al. Prostaglandin E2 increases fibroblast gene-specific and global DNA methylation via increased DNA methyltransferase expression. FASEB J 2012; 26:3703 - 14; http://dx.doi.org/10.1096/fj.11-203323; PMID: 22645246
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature 1986; 321:209 - 13; http://dx.doi.org/10.1038/321209a0; PMID: 2423876
- Chavey C, Mühlbauer M, Bossard C, Freund A, Durand S, Jorgensen C, et al. Interleukin-8 expression is regulated by histone deacetylases through the nuclear factor-kappaB pathway in breast cancer. Mol Pharmacol 2008; 74:1359 - 66; http://dx.doi.org/10.1124/mol.108.047332; PMID: 18669446
- Böcker U, Nebe T, Herweck F, Holt L, Panja A, Jobin C, et al. Butyrate modulates intestinal epithelial cell-mediated neutrophil migration. Clin Exp Immunol 2003; 131:53 - 60; http://dx.doi.org/10.1046/j.1365-2249.2003.02056.x; PMID: 12519386
- Ito K, Lim S, Caramori G, Chung KF, Barnes PJ, Adcock IM. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J 2001; 15:1110 - 2; PMID: 11292684
- Wen X, Wu GD. Evidence for epigenetic mechanisms that silence both basal and immune-stimulated transcription of the IL-8 gene. J Immunol 2001; 166:7290 - 9; PMID: 11390479
- Adam E, Quivy V, Bex F, Chariot A, Collette Y, Vanhulle C, et al. Potentiation of tumor necrosis factor-induced NF-kappa B activation by deacetylase inhibitors is associated with a delayed cytoplasmic reappearance of I kappa B alpha. Mol Cell Biol 2003; 23:6200 - 9; http://dx.doi.org/10.1128/MCB.23.17.6200-6209.2003; PMID: 12917341
- Ashburner BP, Westerheide SD, Baldwin AS Jr.. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 2001; 21:7065 - 77; http://dx.doi.org/10.1128/MCB.21.20.7065-7077.2001; PMID: 11564889
- Iwata K, Tomita K, Sano H, Fujii Y, Yamasaki A, Shimizu E. Trichostatin A, a histone deacetylase inhibitor, down-regulates interleukin-12 transcription in SV-40-transformed lung epithelial cells. Cell Immunol 2002; 218:26 - 33; http://dx.doi.org/10.1016/S0008-8749(02)00523-3; PMID: 12470611
- Hoshimoto A, Suzuki Y, Katsuno T, Nakajima H, Saito Y. Caprylic acid and medium-chain triglycerides inhibit IL-8 gene transcription in Caco-2 cells: comparison with the potent histone deacetylase inhibitor trichostatin A. Br J Pharmacol 2002; 136:280 - 6; http://dx.doi.org/10.1038/sj.bjp.0704719; PMID: 12010777
- Klug M, Heinz S, Gebhard C, Schwarzfischer L, Krause SW, Andreesen R, et al. Active DNA demethylation in human postmitotic cells correlates with activating histone modifications, but not transcription levels. Genome Biol 2010; 11:R63; http://dx.doi.org/10.1186/gb-2010-11-6-r63; PMID: 20565882
- Chiurazzi P, Pomponi MG, Pietrobono R, Bakker CE, Neri G, Oostra BA. Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum Mol Genet 1999; 8:2317 - 23; http://dx.doi.org/10.1093/hmg/8.12.2317; PMID: 10545613
- D’Alessio AC, Weaver IC, Szyf M. Acetylation-induced transcription is required for active DNA demethylation in methylation-silenced genes. Mol Cell Biol 2007; 27:7462 - 74; http://dx.doi.org/10.1128/MCB.01120-07; PMID: 17709385
- Hadizadeh S, King DN, Shah S, Sewer MB. Sphingosine-1-phosphate regulates the expression of the liver receptor homologue-1. Mol Cell Endocrinol 2008; 283:104 - 13; http://dx.doi.org/10.1016/j.mce.2007.11.030; PMID: 18191017
- Sun HS, Hsiao KY, Hsu CC, Wu MH, Tsai SJ. Transactivation of steroidogenic acute regulatory protein in human endometriotic stromalcells is mediated by the prostaglandin EP2 receptor. Endocrinology 2003; 144:3934 - 42; http://dx.doi.org/10.1210/en.2003-0289; PMID: 12933667
- Lopes FL, Desmarais J, Ledoux S, Gévry NY, Lefevre P, Murphy BD. Transcriptional regulation of uterine vascular endothelial growth factor during early gestation in a carnivore model, Mustela vison. J Biol Chem 2006; 281:24602 - 11; http://dx.doi.org/10.1074/jbc.M602146200; PMID: 16790435
- Raychaudhuri B, Vogelbaum MA. IL-8 is a mediator of NF-κB induced invasion by gliomas. J Neurooncol 2011; 101:227 - 35; http://dx.doi.org/10.1007/s11060-010-0261-2; PMID: 20577780
- Auf G, Jabouille A, Guérit S, Pineau R, Delugin M, Bouchecareilh M, et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc Natl Acad Sci U S A 2010; 107:15553 - 8; http://dx.doi.org/10.1073/pnas.0914072107; PMID: 20702765
- Piperi C, Samaras V, Levidou G, Kavantzas N, Boviatsis E, Petraki K, et al. Prognostic significance of IL-8-STAT-3 pathway in astrocytomas: correlation with IL-6, VEGF and microvessel morphometry. Cytokine 2011; 55:387 - 95; http://dx.doi.org/10.1016/j.cyto.2011.05.012; PMID: 21684758
- Mai A, Massa S, Pezzi R, Rotili D, Loidl P, Brosch G. Discovery of (aryloxopropenyl)pyrrolyl hydroxyamides as selective inhibitors of class IIa histone deacetylase homologue HD1-A. J Med Chem 2003; 46:4826 - 9; http://dx.doi.org/10.1021/jm034167p; PMID: 14584932
- Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, et al. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J Med Chem 2005; 48:3344 - 53; http://dx.doi.org/10.1021/jm049002a; PMID: 15857140